



INTRODUCCION
Entre 1786 y 1788 Francisco de Goya y Lucientes pintó para la Real Fábrica de Tapices el cuadro que tituló Los pobres en la fuente, actualmente en el Museo del Prado (fig. 1). El niño de la derecha, que pudiera guardar sus manos para calentarlas entre el pecho y la chaqueta, tiene unos rasgos especiales en la cara -aunque cabe que estuviera encogido por el frío o enfadado-, y los hombros se ven redondeados. También parece de baja estatura, pues el cenit de su cabeza llega a la altura del ombligo de la mujer adulta, pero al desconocer su edad real, sólo se logra elucubrar que acaso era hipocrecido. Quedándonos con los rasgos faciales, evidentes de una disostosis facial bilateral, se puede sospechar la posibilidad de un síndrome de Noonan, que tras el síndrome de Down, es la causa genética más común de hipocrecimiento e hipogonadismo asociado.

Figura 1. Francisco de Goya y Lucientes: Los pobres en la fuente, 1786-1788 para la Real Fábrica de Tapices. Museo del Prado. Madrid, España.
El llamado síndrome de Noonan, descrito por Noonan y Ehmke1 en 1963, de gran polimorfismo expresivo y en el que el signo guía más importante, para el diagnóstico, lo constituyen los peculiares rasgos faciales, a los que se asocian al menos y en combinación variable: estatura corta, deformidades esqueléticas, cardiopatías congénitas, defectos gonadales y en, algunos casos, déficit mental. Todo ello en un fenotipo que va cambiando evolutivamente, con una fórmula citogenética normal. Algunos autores, anteriores a la Jacqueline Noonan1, habían publicado pacientes con características similares, planteándolos como diagnóstico diferencial del síndrome de Turner, con el que comparte varios rasgos y con el distingo de su normalidad cromosómica en ambos sexos, a partir de la descripción citogenética con fórmula 45/X0 para el mencionado síndrome de Turner. Así se le denominaba a aquel "fenotipo Turner con cariotipo normal", "Turner masculino", "síndrome de Ullrich", "fenotipo Turner familiar"2. La virtud de Jacqueline Noonan, además de indicar los signos clínicos mayores, estuvo en observar, en 1968, que la cardiopatía más frecuente era la estenosis pulmonar, frente a la cardiopatía que se presenta en el estado 45/X0 que es la coartación de la aorta3.
CASO CLÍNICO
Datos generales y antecedentes
Paciente de 10 años, sexo masculino, mestizo, procedente del área de Suramérica, que motiva su consulta la recurrencia de bolsas escrotales vacías y corta estatura. Como antecedentes patológicos personales prenatales se encontró la amenaza de aborto, con nacimiento a término y escrotos vacíos, que no fueron resueltos hasta los 6 años con respuesta fallida a orquidopexia en un período de 1 año. Como antecedentes posnatales se constata agenesia renal izquierda.
Su historia familiar por línea paterna muestra estatura corta y hermano con criptorquidia bilateral.
Examen físico
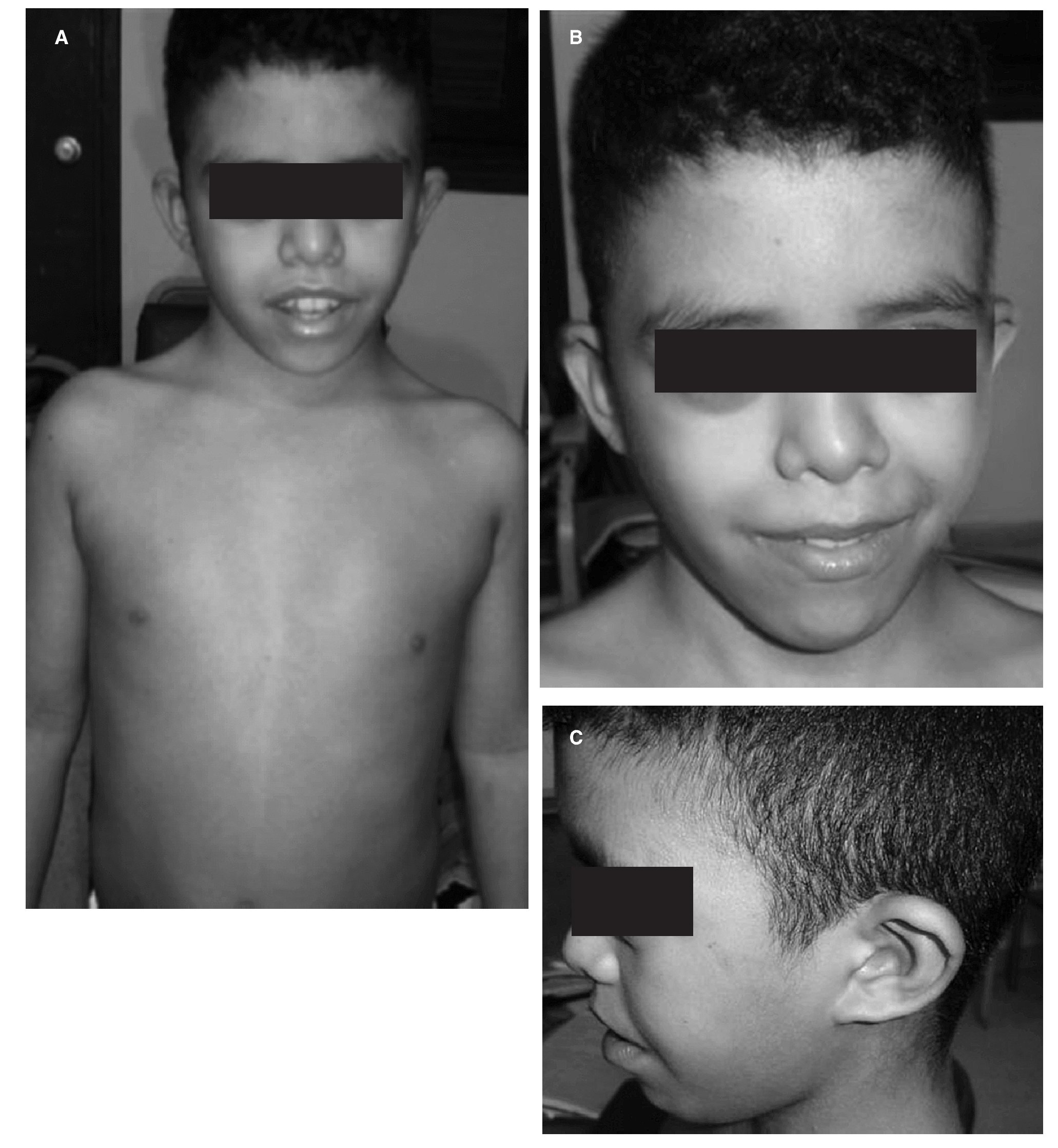
La disarmonía corporal delataba rasgos de polimorfismos, con facies característica: hipertelorismo, epicanto, hendidura palpebral antimongoloide con ptosis, microngnatia e implantación baja de las orejas y cabello en tridente. El pterigium colli resalta el cuello, sin tiroides palpable. Hay presencia de teletelia. Entre las anomalías esqueléticas, se encontraron escoliosis a doble curva, cubito valgo, clinodactilia y acortamiento del cuarto metacarpiano. La evaluación endocrina muestra, paciente prepuberal, con testes en posición no escrotal, de localización canalicular. Desde el punto de vista antropométrico, el peso de 24 kg lo sitúa -4 desviaciones estándar (DE) para su edad, correspondiente a un peso de un niño de 8 años. Su talla de 122,5 cm corresponde a la de un niño de 6 años, ubicada -6 DE para su edad, con una relación P/T mayor 97 pc. Su perímetro cefálico de 53 cm denota macrocránea. La brazada en 128,5 cm como signo indirecto de hipogonadismo y relación SS/SI mayor de 2 (fig. 2).
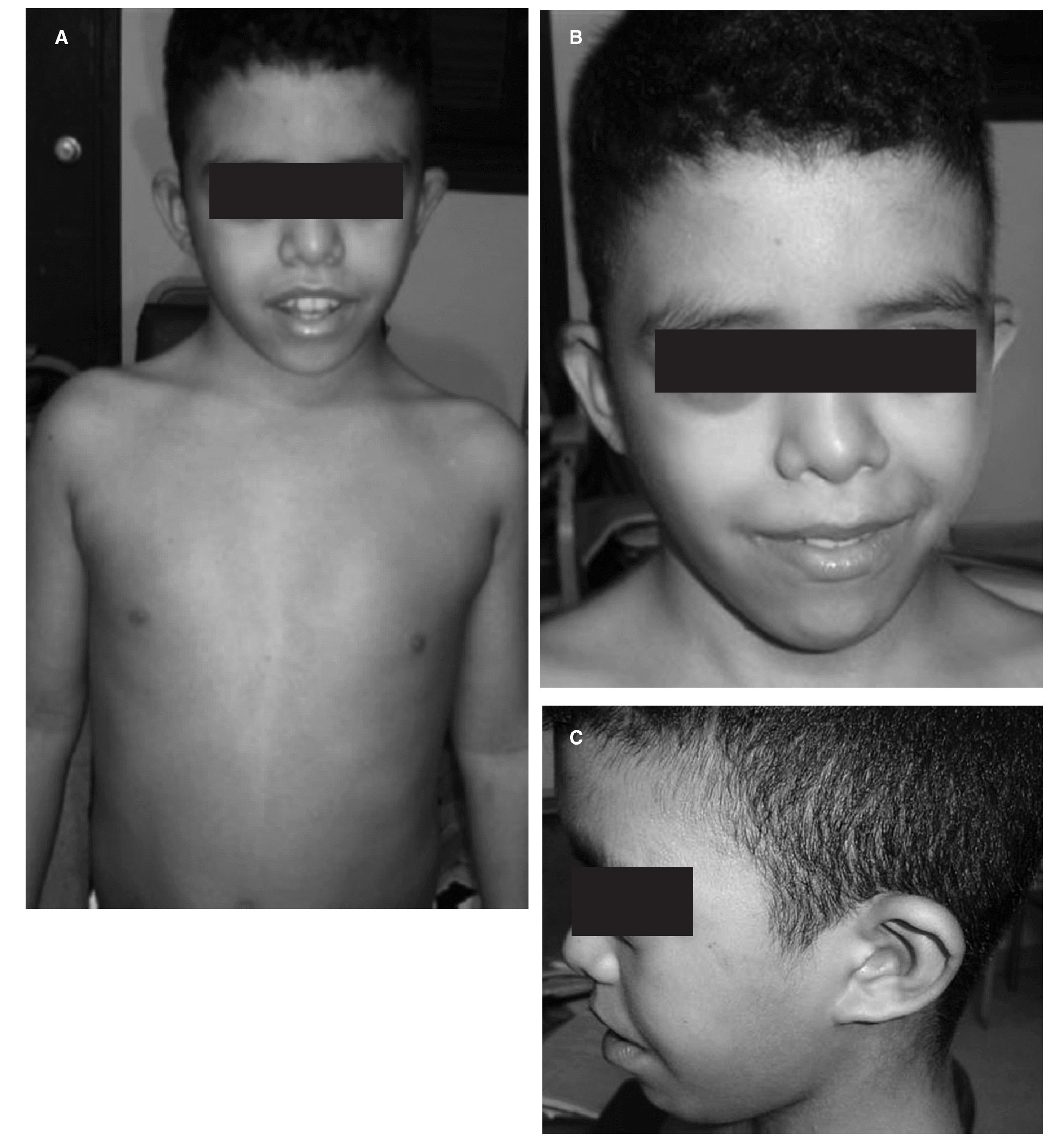
Figura 2. Disarmonía corporal mostrando rasgos de polimorfismos (A), con facies característica: hipertelorismo, epicanto, hendidura palpebral antimongoloide con ptosis, microngnatia (B) e implantación baja de las orejas y cabello en tridente (C).
Bioquímica
Eje gonadotrópico con valores elevados (hormona foliculostimulante [FSH]: 4,4 mU/ml y hormona luteinizante [LH]: 9,8 mU/ml), bajos valores de testosterona basal (12,4 nmol/l) sin respuesta al test de estimulación con gonadotropina coriónica humana (HCG) (14,1 nmol/l). Sin disfunción tiroidea. Pruebas dinámicas de HGH sin respuesta al estímulo de la clonidina y a la hipoglucemia inducida por insulina, con un patrón de deficiencia total.
Genética
Cariotipo-46XY.
Imagenología
La ultrasonografía abdominal fue normal. El Doppler testicular describió un tejido testicular situado en localización canalicular, entre anillos inguinales interno y externo bilateral. El ecocardiograma fue normal. El mapa radiográfico reveló una edad ósea retardada correspondiente a 6 años.
Histología
La biopsia testicular muestra ligera disminución del tamaño de los túbulos seminíferos y numero de espermatogonias, sin esclerosis, discreto aumento del tejido peritubular y ligera hiperplasia de células de Leydig.
Neurocognición
Test psicométrico WISCH informa de un bajo cociente de inteligencia (CI = 68).
Terapéutica
El tratamiento se encamina a la corrección quirúrgica del defecto testicular, y se realiza orquidopexia bilateral exentas de complicaciones y resultados satisfactorios, con seguimiento programado para evaluar el futuro puberal. Se instala terapia de reemplazo hormonal sobre el eje somatotropo con Norditropin a las dosis establecidas.
DISCUSIÓN
El síndrome de Noonan afecta al menos a 1 de cada 2.500 niños2. A diferencia del síndrome de Turner, los afectados no tienen alteraciones de las estructuras que contienen la información genética. Diferentes estudios detectaron familias donde el síndrome aparecía en varios miembros, con una transmisión vertical, con rasgos diferentes de unos a otros e incluso con generaciones saltadas (dominancia irregular), pero con un predominio de herencia por vía materna. Esto estableció un patrón de herencia autosómica dominante, lo que significa que en estos casos sería necesaria la presencia de la mutación en los genes de uno de los progenitores, y que ésta sería transmitida a los hijos con un 50% de probabilidad, con solo un 14% de expresión en su forma grave. El hecho de que algunos niños no tengan un padre con el síndrome de Noonan refleja la posibilidad de una aparición esporádica, lo que presume la presencia de una nueva mutación, no presente en los genes de los padres4-8.
Jamieson et al6 identificaron el locus donde se ubica el gen que condiciona el fenotipo, de al menos, un gran porcentaje de personas con síndrome de Noonan, situándolo en la posición 12q24. En este emplazamiento del mapa genético se encuentra el primer gen específico identificado como posible responsable del síndrome, denominado PTPN11, el cual no puede explicar aún todos los casos. Existen niños nacidos de parejas consanguíneas, en los que el cuadro clínico no parece diferir llamativamente de la forma dominante, salvo que tienen alta frecuencia de miocardiopatía hipertrófica obstructiva manifestada muy precozmente por lo que se mantiene la búsqueda de otros posibles genes implicados en su génesis7,8.
Este trastorno produce desarrollo anormal de múltiples partes del cuerpo y lo caracterizan una serie de elementos físicos que pueden variar ampliamente en rango y severidad según los casos. El fenotipo es muy característico, junto a la baja talla con retraso de la edad ósea destacan el pterigium colli y las alteraciones craneofaciales: hipertelorismo, raíz nasal amplia, maxilar estrecho, oblicuidad antimongoloide de las hendiduras palpebrales con ptosis, boca "de carpa", pabellones auriculares de implantación baja, paladar ojival y maloclusión dentaria. Es frecuente el tórax escavado y menos frecuente las alteraciones vertebrales, como escoliosis. Al igual que en el síndrome de Turner puede estar presente el cubito valgo y la teletelia, así como diversas anomalías asociadas: renales, cutáneas (nevus), oculares y linfáticas (linfedemas)9,10.
El estudio endocrino pone de manifiesto una insuficiencia gonadal primaria y elevación de la concentración plasmática de gonadotropinas en la edad puberal. Las alteraciones gonadales propias del sexo masculino consisten en testes pequeños y criptorquidia, en ocasiones con hipogonadismo hipergonadotropo. Microscópicamente puede demostrarse disminución del diámetro tubular y del índice de fertilidad en los pacientes prepuberales. Generalmente se presenta un retardo en la pubertad con afectación de la espermatogénesis en adultos. En las mujeres se han descritos ovarios de aspectos disgenéticos por laparoscopia con desarrollo deficiente del aparato folicular9,10.
El 70% de los pacientes tienen alteraciones cardíacas diversas; predomina la estenosis pulmonar, la comunicación interauricular y la hipertrofia septal asimétrica. Suelen presentar hipoplasia o aplasia de vasos sanguíneos y linfáticos, alteraciones de las plaquetas y de los factores de la coagulación de la sangre e hipertermia maligna11.
El retardo mental leve en aproximadamente el 25% de los casos puede o no asociarse a hipotiroidismo. Existe pérdida auditiva variable, sin olvidarse de la mayor frecuencia de defectos de refracción. También se han comunicado neurofibromatosis, coagulopatías, tiroiditis autoinmunitaria, hipotiroidismo e hipoparatiroidismo, déficit de hormona de crecimiento, feocromocitoma, ganglioneuroma, schwannoma maligno y otras afecciones. Suelen tener retraso mental moderado, asociado o no a hipotiroidismo. Por lo demás, como cualquier otro niño, pueden darse dificultades para la alimentación como la anorexia, muy vinculada a reflujo gastroesofágico9,10,12.
El diagnóstico del síndrome de Noonan es fundamentalmente clínico y se descubren como tales a todos aquello que cumplan las características fenotípicas que definen este síndrome. Un sencillo examen físico puede mostrar cualquiera de las alteraciones faciales o corporales del mismo. El diagnóstico prenatal solamente cabe en forma de sospecha ante la presencia de higroma quístico nucal o linfedema, cuando el cariotipo es 46, XX o 46, XY. No hay criterios ultrasonográficos para la identificación precoz9,10,12.
El diagnóstico diferencial debe hacerse, en las niñas fundamentalmente, con el síndrome de Turner y en ambos sexos es preciso excluir sobre todo la embriofetopatía alcohólica y por primidona, amén de otros cuadros con algunos rasgos comunes como el síndrome de Aarskog o la trisomía del brazo corto del cromosoma 84-8.
No hay un tratamiento único y específico para el síndrome de Noonan. El plan terapéutico se centra y ejecuta según los problemas que se presentan. Tras el diagnóstico neonatal fenotípico externo, se impone la evaluación cardiológica como primera medida a seguir. Se refiere que hasta un 6% fallece cada año por arritmia11. Luego hay que centrarse en la observancia evolutiva del crecimiento pondoestatural y del desarrollo psicomotor9,10,12.
El uso de la hormona de crecimiento es eficaz a corto plazo aumentando la velocidad de crecimiento aunque sin que al parecer se incremente de forma ostensible la talla final: media de + 3,1 cm (-1,1 a 6,5 cm). No se ha constatado riesgo de cardiomiopatía hipertrófica bajo el tratamiento en el curso de 3 años de terapéutica12.
En la pubertad, máxime los varones que tuvieron criptorquidia bilateral, han de ser evaluados periódicamente, tanto en su desarrollo puberal como las determinaciones hormonales para verificar la posibilidad de hipofunción. El tratamiento con de testosterona de depósito cada 21 días o parche diario, ha de ser individualizado, para un evitar la osteoporosis, y no se estimule la libido, potencialmente peligrosa, si no se controla con una inteligencia y educación suficientes9,10,12.
El pronóstico esperado depende de la extensión y gravedad de los síntomas existentes. Los pacientes pueden llevar una vida normal y, si se presenta retraso mental, generalmente es leve.
En las personas con antecedentes familiares de síndrome de Noonan se impone el consejo genético al pretender descendencia. La prevención de las complicaciones, tal como enfermedad cardíaca, depende de la detección a tiempo y el cuidado continuo de un cardiólogo12.
CONCLUSIONES
Tras presentar y describir este caso clínico de síndrome de Noonan en un varón, concluimos que resulta de suma importancia, en la evaluación de los trastornos gonadales en la niñez, tener en cuenta los defectos de naturaleza genética. A pesar de solucionar uno de los trastornos de la gónada, resulta trascendente la continuidad en la evaluación del futuro puberal en estas circunstancias. Cobra envergadura el modelo de corrección de otros defectos hormonales importantes, involucrados en este síndrome, sobre todo los trastornos del crecimiento y evaluar cautelosamente el desarrollo neurocognitivo.
AGRADECIMIENTOS
Al Dr. Pedro Ramón Gutiérrez Hernández, Editor-Jefe de la REVISTA INTERNACIONAL DE ANDROLOGÍA.
SALUD SEXUAL Y REPRODUCTIVA, por su ánimo e insistencia en la publicación de este caso clínico, expresado en las sugerencias y correcciones de éste.
Correspondencia:
Dra. C. Pérez Gesen.
Instituto Nacional de Endocrinología. Zapata y Plaza de la Revolución. 10400 Ciudad de La Habana. Cuba.
Correo electrónico: cecilia.gesen@informed.sld.cu