


La sepsis es un síndrome complejo y devastador que persiste como una causa importante de morbilidad y mortalidad, entre los pacientes críticamente enfermos. La prevalencia de sepsis y choque séptico ha ido en incremento significativo en las últimas dos décadas. A pesar del desarrollo de investigación básica y numerosos ensayos clínicos dedicados al problema, no se han logrado avances notables en el desarrollo de terapias vanguardistas y eficaces para su manejo. Los trastornos fisiológicos inducidos por la sepsis son en gran parte debidos a la respuesta del huésped a los microorganismos invasores, en contraste con los efectos directos del propio microorganismo.
La sepsis entendida como la respuesta inflamatoria sistémica a un proceso infeccioso, se caracteriza por una disrregulación en la síntesis de citocinas proinflamatorias. Pese a que la producción de citocinas proinflamatorias es indispensable normalmente para proteger contra los patógenos y promover la reparación de los tejidos, la producción disrregulada y prolongada de éstas, puede desencadenar una cascada inflamatoria sistémica mediada por quimiocinas, aminas vasoactivas, los sistemas del complemento y de la coagulación y especies reactivas de oxígeno, entre otros. Estos mediadores conducen de manera colectiva a la insuficiencia orgánica múltiple, y en última instancia, a la muerte.
Sepsis is a complex and devastating syndrome that remains as an important cause of morbidity and mortality in critically ill patients. The prevalence of sepsis and septic shock have increased steadily over the past two decades. Despite basic research and numerous clinical trials there has been no real progress in its effective management. The physiologic derangements induced by sepsis are mainly induced by the host's response to the invading microorganisms, in contrast to the direct effects of the invading microorganism by itself.
Sepsis, understood as the systemic inflammatory response to an infectious process is characterized by a dis-regulation in the synthesis of proinflammatory cytokines, synthesis that is an indispensable and normal step to protect the host against invading microorganisms and promote tissue repair. The prolonged and dis-regulated production of these cytokines can unleash a systemic inflammatory cascade mediated by chemokines, vasoactive amines, the complement and coagulation systems and reactive species of oxygen, among others. These mediators concur to induce multiple organ failure and, eventually, death.
> Antecedentes
Las manifestaciones clínicas de la sepsis ya eran conocidas por Hipócrates (460-377 a. C.), quien introdujo el término "herida putrefacta". En Persia, el padre de la medicina Ibn Sina (también conocido como Avicena, 980-1037 d. C.), observó que la sepsis era habitualmente acompañada por fiebre. Sin embargo, no fue hasta mediados del siglo XVIII, que Louis Pasteur relacionó la putrefacción de las sustancias con bacterias y microrganismos. Ignaz Semmelweis observó un hecho significativo al implementar medidas de higiene y la consecuente disminución de la mortalidad, en mujeres en puerperio. En 1914, Hugo Shottmüller presentó los fundamentos para la definición moderna de sepsis y fue el primero en describir la presencia de la infección como parte fundamental de la enfermedad. En décadas posteriores, las ideas de Lewis Thomas llevaron a un cambio radical en el entendimiento de la sepsis por su teoría de "... Es la respuesta del hospedador...la que provoca la enfermedad...". Esta teoría deriva de un gran número de estudios experimentales, que eventualmente fueron cambiando la conceptualización de la sepsis que hasta entonces se tenía. Finalmente, el concepto actual fue introducido a la práctica clínica, cuando Roger Bone definió a la sepsis como un síndrome de respuesta inflamatoria sistémica (SRIS), que puede ocurrir durante la evolución de una infección.1,2
En el pasado, se pensaba que la sepsis iniciaba como una respuesta del sistema inmunitario innato con la consecuente activación de la cascada proinflamatoria en respuesta a la infección secundaria por un agente patógeno o tejido dañado del hospedador (quemaduras o trauma).2 La activación del sistema inmune innato en el que destacan las células dendríticas, macrófagos, neutrófilos, el complemento y el sistema de la coagulación, después de la exposición al estímulo inicial, generan una respuesta inflamatoria abrumadora, resultando en la principal característica de la sepsis.
Este ambiente proinflamatorio induce la liberación de mediadores secundarios potentes (factores lipídicos y especies reactivas de oxígeno), que amplifican el proceso inflamatorio. La disfunción de los mecanismos reguladores de la sepsis resulta en la disminución del control inflamatorio responsable del daño exagerado a los tejidos y órganos del hospedador.
El fracaso de las terapias antiinflamatorias para el control de la sepsis empleados en ensayos clínicos previos, han planteado el cuestionamiento de si la mortalidad en sepsis es en realidad derivada de la respuesta inflamatoria no controlada.3 Pese a que algunos pacientes mueren durante la fase hiperinflamatoria inicial de la sepsis, la mayoría de los enfermos con sepsis grave y choque séptico sucumben en fases posteriores de la misma y asociado con un prolongado estado de inmunosupresión.4,5 Este hecho ocurre particular en los neutrófilos, los cuales pueden verse sometidos a una "parálisis inmune" durante la sepsis grave, lo que implica la parálisis completa de importantes vías de señalización intracelulares y disfunción del sistema inmune adaptativo, el cual es también un importante factor contribuyente a la inmunosupresión que se observa en etapas tardías de la sepsis.
Está demostrado que gran parte de la respuesta inflamatoria está orquestada por las células T, principalmente las CD4+, siendo la diferenciación de éstas entre el complejo TH1 y TH2, lo que definirá el perfil de citocinas liberadas.
Durante la sepsis, la respuesta inmune adaptativa desvía la respuesta inicial de células TH1 (caracterizado por la producción de interferón gamma (IFNγ) y la interleucina-12 (IL-12) a una respuesta de células TH2 (caracterizado por la producción de IL-4, IL-5, IL-10 e IL-13), lo que puede provocar una profunda inmunosupresión. La interacción celular TH1-TH2 y el paradigma celular se ha ampliado recientemente con el descubrimiento de las células TH17, un subconjunto celular TH que produce IL-17. Se ha considerado a las células TH17 como parte importante de la inmunidad contra los microorganismos que no son eliminados por la respuesta inmune, mediada a través de las células TH1 y TH2.6
El incremento de la apoptosis en los linfocitos y células dendríticas (CD), también contribuye en buena medida a la supresión de la respuesta inmune durante la sepsis. Además de causar una marcada disminución en el número de células, la apoptosis de los linfocitos y CD generan inmunoparálisis a través de los efectos inmunosupresores de las células apoptóticas. En contraste, los linfocitos de pacientes en riesgo, la apoptosis de los macrófagos y los neutrófilos parece no estar afectada e inclusive disminuida durante la sepsis.7,8 Considerando que el aumento de la apoptosis de los linfocitos y células dendríticas culmina en un estado de inmunosupresión severa, lo cual coloca a los pacientes en un alto riesgo de adquirir infecciones de carácter nosocomial, la disminución en la apoptosis de los neutrófilos, incrementa el daño causado debido a su actividad proinflamatoria. Datos recientes indican que se requiere de la supresión de las células T mediada en la fase temprana de la respuesta de la inmunidad innata, para poder minimizar el daño a los tejidos del hospedador y maximizar la respuesta de defensa.9
En la actualidad, existe evidencia de que la sepsis es una condición que afecta no sólo al sistema inmune sino también a otros sistemas biológicos, tales como el sistema de coagulación y el sistema nervioso autónomo (SNA).10-12
1. Respuesta inflamatoria
Las células inmunes expresan un conjunto de receptores conocidos como receptores de patrones de reconocimiento (RPR), que rápidamente inician la respuesta de defensa del hospedador después de haber detectado la presencia de daño tisular o infección microbiana.
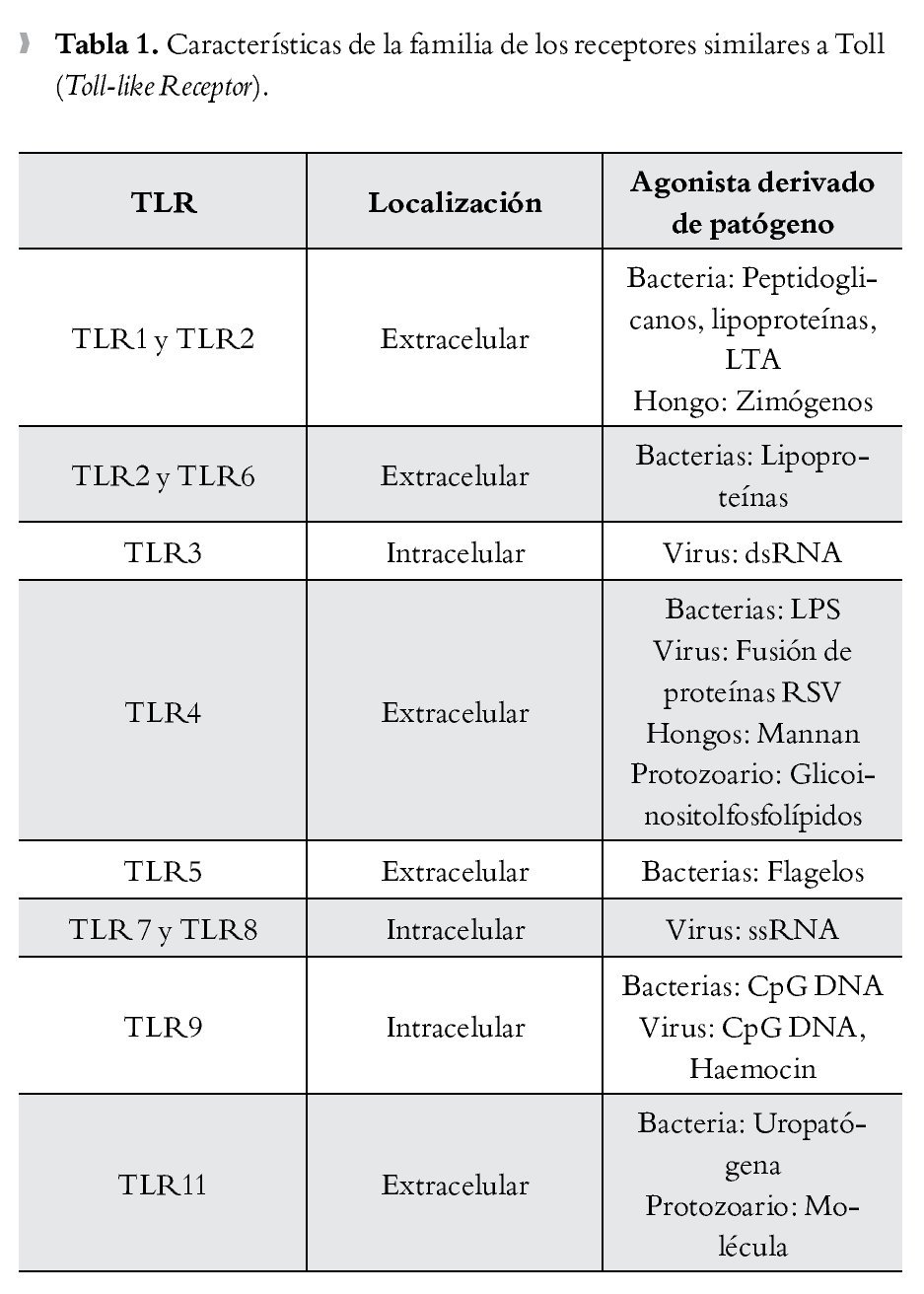
La presencia de una infección microbiana se detecta mediante el reconocimiento conservado de patrones moleculares asociados a patógenos (PMAPs), que se expresan tanto en microorganismos invasores patógenos como en los inocuos. Por el contrario, el reconocimiento inmunológico del tejido dañado está mediado por proteínas intracelulares o mediadores que se liberan de las células que mueren. Se denominan a estas proteínas "alarminas", que junto con los PMAPs, se conocen como patrones moleculares asociados a daño (PMADs). Los receptores de tipo Toll (TLR), que son una subfamilia de los RPRs, han surgido como receptores cruciales para el reconocimiento de PMADs y en la iniciación de la respuesta inflamatoria13 (Tabla 1).
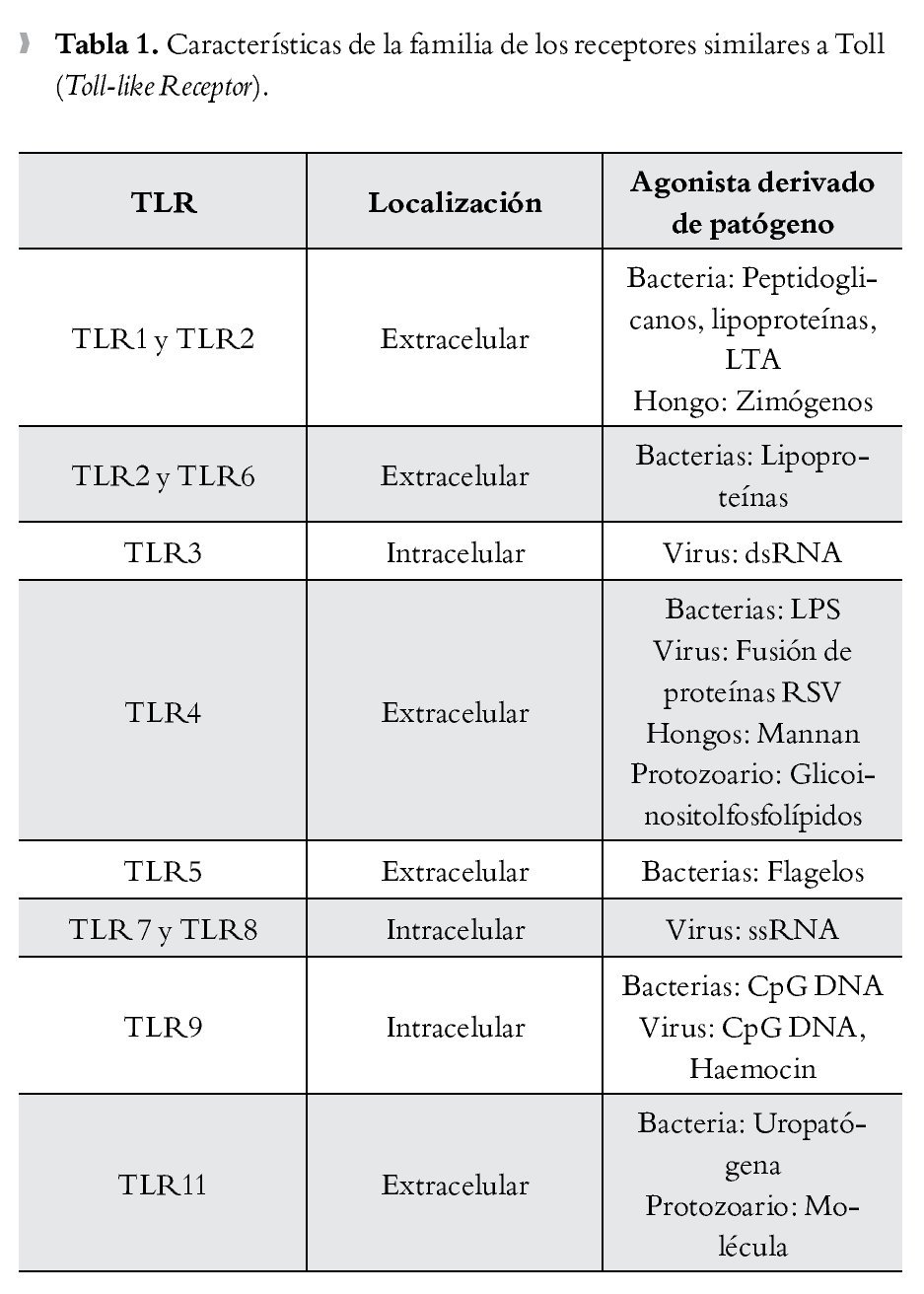
Durante la sepsis, hay una activación sistémica de la respuesta inmunológica debido a la liberación de altas concentraciones de PMADs procedentes tanto de los microorganismos invasores como de los tejidos dañados, favoreciendo la estimulación de las células efectoras de la inmunidad innata. Como resultado, la sepsis se acompaña de una marcada y desequilibrada respuesta mediada por citocinas (conocido como "tormenta de citoquinas"), que convierte esta respuesta fisiológica de defensa en un estado inflamatorio excesivo y perjudicial.
TLR4
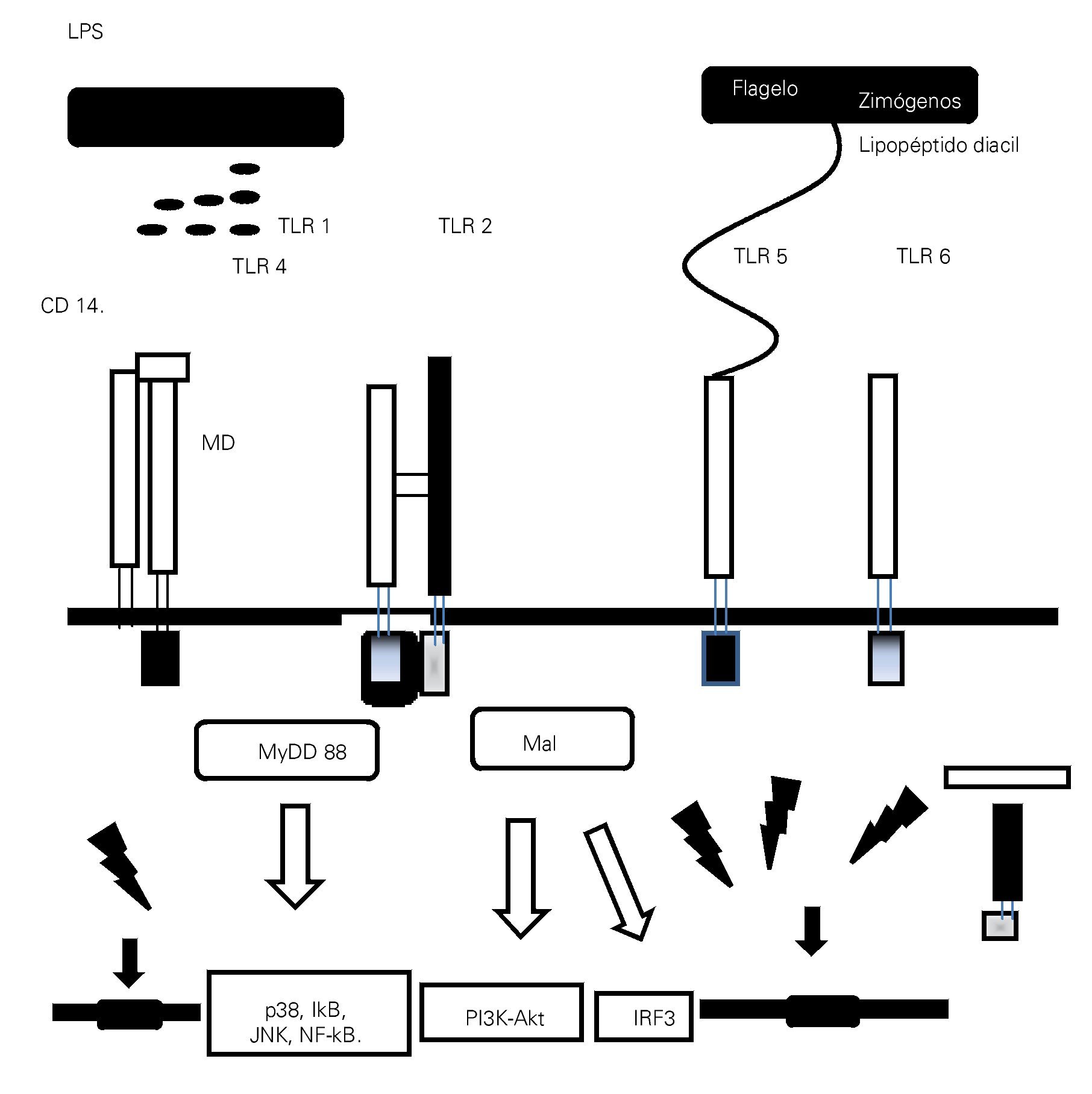
El reconocimiento de lipopolisacáridos (LPS) mediada por TLR4, un PMAPs bien caracterizado ubicado en la membrana externa de las bacterias Gram negativas, se piensa es un importante disparador de la respuesta inflamatoria en sepsis.14,15 TLR4 forma un receptor complejo con CD14 y MD2, este último también juega un importante papel en el reconocimiento de LPS.16,17 Además de los LPS, se han descrito diversos ligandos endógenos para TLR4, incluyendo a las proteínas del complejo de alta movilidad Box-1 (Toll-like Receptor).
(High Mobility Group Box 1, HMGB1), las cuales son un importante mediador durante la fase tardía de la sepsis.18 Se ha postulado que la diafonía (cross talk) que se produce entre TLR4 y el sistema del complemento, está implicada en la iniciación de la respuesta inflamatoria en sepsis.19,20 La anafilatoxina C5a del complemento regula de manera negativa la respuesta mediada por TLR4.19 La extensión del efecto regulador del complemento en la producción de citocinas mediadas por TLR4, correlaciona con el nivel de los productos de la activación del complemento y, a su vez, las citocinas que son inducidas por la activación de TLR4 regulan la expresión del complemento a través de los receptores de la anafilotoxina C5AR y C3AR.20,21 El hallazgo de que la activación de TLR-4 en las plaquetas inicia la formación de trampas extracelulares en los neutrófilos para "entrampar" bacterias dentro de la circulación, demuestra una vez más la intrincada interacción entre la inmunidad innata y el sistema de coagulación en la fisiopatogenia de la sepsis.22 Debido a su importante papel en la iniciación de la respuesta inflamatoria, TLR-4 resulta en un potencial blanco terapéutico en el manejo de la sepsis (Figura 1).
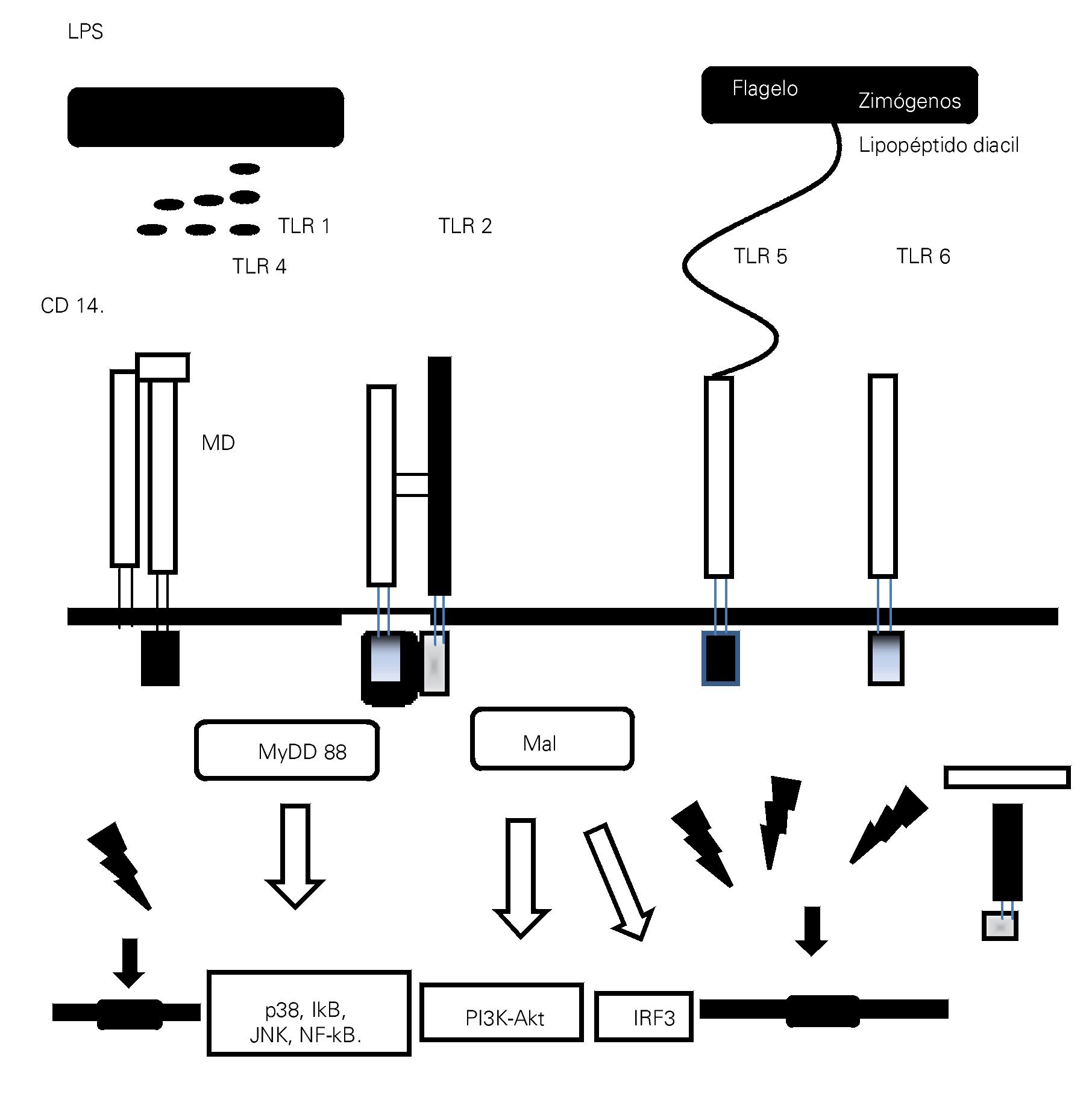
> Figura 1. Esquema que representa la interacción entre los patrones moleculares asociados a patógenos (PMAPs) y a daño (PMADs) con los receptores de patrones de reconocimiento (RPRs), como la familia de los receptores similares a Toll (TLRs), con activación de las vías de señalización citoplasmáticas y nucleares encargadas de mediar la configuración de la respuesta inmunológica en proinflamatoria/antiinflamatoria.
Ejes centrales nocivos en la sepsis
Es a partir del conocimiento de que los mediadores de la inflamación (no sólo los microorganismos invasores) están involucrados en la fisiopatogenia de la sepsis, se han abierto nuevas líneas de investigación sobre los mecanismos fisiopatológicos de la inflamación. Diferentes mediadores se han relacionado con la fisiopatogenia de la sepsis, algunos de los cuales, pueden ser considerados como ejes centrales de la compleja red inflamatoria. Aunque difieren en términos de su procedencia, la cinética de liberación y la fase de la sepsis en la cual predominan estos ejes centrales, pueden ejercer efectos pleiotrópicos al interconectar diversas vías de la respuesta inmune.
C5a
Como parte de la respuesta inmune innata, el sistema del complemento se activa durante la etapa temprana de la sepsis, lo que genera grandes cantidades de anafilotoxina C5a. En altas concentraciones C5a tiene múltiples efectos nocivos. En consecuencia C5a actúa como mediador central de la sepsis mediante la modulación de otros sistemas (cascada de la coagulación, respuestas mediadas por TLR4 y liberación de citocinas tales como el factor inhibidor de la migración de macrófagos MIF y HMGB1).10,19,23-26.
MIF
Es una de las primeras citocinas aisladas, tiene un papel fundamental en la regulación de la respuesta inflamatoria sistémica y local. Las endobacterias, exotoxinas, mediadores proinflamatorios, así como el TNFa y C5a, a diferencia de otras citocinas, se expresa constitutivamente en los leucocitos y se almacena en el compartimento intracelular. Después de su secreción, el MIF funciona como una citocina proinflamatoria clásica y promueve la respuesta inmune innata y adaptativa por activación de macrófagos y células T.27 La actividad proinflamatoria de MIF está mediada por actividad de su automerasa, que está codificada por un dominio que contiene un sitio catalítico conservado evolutivamente.28 Además de mediar su efecto proinflamatorio, el MIF también induce y amplifica la producción de otras citocinas proinflamatorias y regula positivamente la expresión de TLR4 por fagocitos.27 En altas concentraciones el MIF evita la apoptosis dependiente de p53 de macrófagos activados, que son responsables de la respuesta inflamatoria sostenida. Sin embargo, los mecanismos exactos por los cuales el MIF ejerce sus efectos biológicos en el contexto de la inflamación no están del todo elucidados. Aunque el MIF activa vías de señalización intracelulares después de su endocitosis, el complejo receptor CD74 también ha sido descrito que funciona como un receptor de MIF, a través del cual traduce señales mediante CD44.29
El MIF es único entre las citocinas, ya que vincula el sistema inmune con el endocrino. En respuesta al estrés, el MIF es secretado por el hipotálamo, adenohipófisis y glándula suprarrenal.27,30 Es importante destacar que el MIF antagoniza y anula el efecto anti-inflamatorio que podrían tener relevancia para el empleo terapéutico de los corticoesteroides en sepsis.27
Los corticoesteroides endógenos inducen la liberación de MIF a partir de células del sistema inmune, por lo que el efecto inhibidor del MIF en la acción de los corticoesteroides es mediante retroalimentación negativa.31,32 La producción excesiva de MIF es nociva en la fase aguda de la sepsis, ya que los niveles séricos de MIF se han correlacionado con la gravedad de la sepsis. La neutralización o focalización de la actividad del MIF por automerasa es atenuada por la respuesta inflamatoria, mejorando la supervivencia de los modelos experimentales en sepsis.33,34
HMGB1
HMGB1 fue descrito originalmente como un factor de transcripción.35 Después, se reclasificó como una citocina proinflamatoria convirtiéndose en el foco de un gran número de estudios. HMGB1 es expresado por casi todas las células, excepto en aquellas que carecen de núcleo (eritrocitos), y la principal fuente de la HMGB1 en los procesos de inflamación son los macrófagos, monocitos y neutrófilos.36-38 HMGB1 puede ser secretado por células del sistema inmune después de su acetilación a nivel nuclear y la subsiguiente translocación hacia el citoplasma, o ser liberado a partir de células necróticas.37 La secreción activa de HMGB1 es regulada por la activación del factor nuclear kappa-B (FNkB), probablemente a través de mecanismos no transcripcionales, aunque la forma en que esto ocurre no es aún del todo determinada.39
Destaca el hecho de que pese a que las células apoptóticas no son una fuente de HMGB1 extra-celular, éstas provocan que los macrófagos liberen HMGB1 durante la sepsis.40,41 Específicamente el HMGB1 extracelular interactúa con RPRs, incluyendo el receptor para los productos finales de glicación avanzada (RAGE), TLR2 y TLR4. La señalización inducida por HMGB1 posee efectos pleiotrópicos sobre las células del sistema inmune, promueve la inflamación y potencialmente la disrupción por daño sobre las barreras epiteliales.18,37,42 Además de la activación de los RPRs, el HMGB1 incrementa la actividad proinflamatoria de las citocinas (IL-1β) a través de la unión a estos mediadores, lo que apoya la idea de que HMGB1 podría no actuar únicamente como un mediador proinflamatorio, sino también podría funcionar como un transportador de PMAPs.43,44
Aunque HMGB1 se libera a nivel sistémico durante la sepsis, los niveles plasmáticos no necesariamente se correlación con el desenlace o supervivencia.37 En contraste con otras citocinas asociada a la sepsis, el pico de liberación de HMGB1 se produce durante las etapas tardías de la enfermedad, y los niveles de la HMGB1 no siempre disminuyen en los pacientes que se han recuperado de un evento de sepsis.36 Las moléculas derivadas de patógenos y estímulos proinflamatorias (tales como TNFα, IL-1β e IFNγ), inducen la secreción de HMGB1 durante la inflamación.37 La interacción entre C5a y su otro receptor, el receptor 2 similar a C5a (C5L2), también desencadenan la liberación de HMGB1 en sepsis.29
De manera interesante, la secreción de HMGB1 es bajo la influencia del sistema nervioso autonómico (SNA). La activación de la vía colinérgica antiinflamatoria suprime la secreción de HMGB1 de los macrófagos en sepsis, mejorando la supervivencia.39
Dado los efectos pleiotrópicos de HMGB1 en la respuesta inflamatoria y su liberación tardía en la sepsis, HMGB1 podría ser un blanco terapéutico prometedor.
IL-17A
El reciente descubrimiento de la familia de las citocinas IL-17, las cuales han surgido como importantes mediadores de la regulación inmune, han permitido ampliar nuestro conocimiento sobre la interacción entre la respuesta inmune innata y adaptativa. El primer miembro de esta familia descrito fue IL-17, la cual es una citocina proinflamatoria principalmente producida por células TH17. IL-17A también es secretada por otros tipos de células del sistema inmune, incluyendo a los neutrófilos, linfocitos T CD8+, células natural killers, y algunos otros subconjuntos de células TH y células T-γθ. En resumen, IL-17A está involucrada en la mediación de la respuesta proinflamatoria mediante la activación y producción de muchas otras citocinas (IL-1β, IL-6 y TNFα) y proporciona la diafonía entre los linfocitos y fagocitos.45
Se ha demostrado de manera reciente en modelos experimentales de sepsis, que el incremento de los niveles de IL-17A tiene efectos deletéreos. Que la neutralización de IL-17A mejora notablemente la supervivencia, aún e inclusive cuando el tratamiento ha sido administrado de manera tardía hasta 12 horas de haber sido inducida la sepsis de manera experimental. Los efectos protectores del bloqueo de IL-17A se han asociado con una marcada atenuación de la bacteremia y reducción notable de los niveles plasmáticos de citocinas proinflamatorias. De acuerdo con estos datos, la producción in vitro de mediadores proinflamatorios por los macrófagos en respuesta a LPS, es significativamente superior en presencia de IL-17A recombinante. Sin embargo, todavía aún no se sabe si los niveles de IL-17A se incrementan en pacientes con sepsis, o durante qué fase de la sepsis la neutralización de IL-17A sería benéfica en el entorno clínico.
Debido a que la producción de IL-17 es importante para orquestar la respuesta inmune contra infecciones específicas, el bloqueo de la IL-17A en determinadas condiciones podría condicionar más daño que beneficio. Por lo tanto, queda aún por determinar si IL-17A es una blanco útil de intervención terapéutica en la sepsis.46
2. Disrregulación de la cascada plasmática
Complementopatía
El sistema del complemento se puede activar a través de tres vías diferentes que convergen en la generación de las anafilotoxinas C3a y C5a, C4a y el complejo de ataque a la membrana (MAC; también conocido como C5b-C9). En los ensayos clínicos de sepsis, el incremento plasmático de las concentraciones de C3a, C4a y C5a, se han relacionado con un peor pronóstico y sobrevida.47,48 Destaca el hecho de que C3a podría además de actuar como anafilotoxina proinflamatoria, tener propiedades antiinflamatorias. En el modelo murino de sepsis, los ratones con deficiencia de C3AR fueron más susceptibles a desarrollar estado de choque por endotoxemia, el cual fue acompañado de un incremento en la concentración plasmática de citocinas proinflamatorias. La unión de C3a a C3AR puede activar la secreción de hormonas antiinflamatorias a través la glándula hipófisis, lo que podría explicar la capacidad antiinflamatoria de C3a.49,50
Nuevos descubrimientos continúan incrementando nuestro acervo y comprensión acerca de los numerosos efectos nocivos, derivados de la producción excesiva de C5a durante la sepsis. Los efectos derivados de C5a contribuyen al desarrollo de la parálisis inmune, disfunción y falla multiogánica, la apoptosis de timocitos y células de la médula suprarrenal, así como el desequilibrio en el sistema de la coagulación.51-55
Además, C5a está ampliamente relacionada con el desarrollo de cardiomiopatía mediada por sepsis.56
Estudios realizados de forma reciente, confirman el importante papel de C5a en la fisiopatogenia de la sepsis.29 Además de C5AR, C5a puede unirse específicamente a un segundo receptor, C5L2, la función del cual era desconocida hasta hace poco tiempo. Originalmente se postuló que C5L2 funcionaba como un receptor "señuelo" para C5a, compitiendo con C5AR por la unión de C5a, aunque la evidencia reciente indica que C5L2 es un receptor funcional.29,57,58 Actualmente, existe evidencia de que C5AR y C5L2 cooperan para potenciar la respuesta inflamatoria durante la sepsis, aunque cada receptor puede tener roles funcionales específicos y diferentes.29
Coagulopatía
En el ámbito clínico de la sepsis, la disrregulación de la cascada de coagulación destaca por el desarrollo de múltiples complicaciones. La magnitud de la activación de la cascada de la coagulación durante la sepsis puede variar desde un nivel insignificante, hasta la aparición de coagulación intravascular diseminada (CID), inclusive. En la fase inicial de CID, la activación de la trombina da como resultado la formación intra y extravascular de fibrina (proceso conocido como hipercoagulabilidad), seguido por el consumo de factores de la coagulación y disfunción plaquetaria. En la fase tardía de la CID, el acúmulo de fibrina a nivel microvascular se asocia a menudo con el desarrollo de disfunción y falla multiorgánica, este fracaso atribuido a las perturbaciones de la microcirculación.59 La CID desarrolla inflamación y activa la coagulación, e interactúa de manera bidireccional.60 La trombina activada puede promover la activación de diversas vías proinflamatorias, dentro de las que se incluyen la producción de citocinas proinflamatorias (tales como TNFα, IL-1β e IL-6) y la generación de C5a, a su vez que pueden estimular la coagulación.10,61,62 El factor tisular (FT), que es una molécula central para la iniciación de la CID, es expresada por las células endoteliales activadas y por células que no están normalmente expuestas al flujo sanguíneo, tales como las células subendoteliales, fibroblastos y también por células inmunes circulantes. En sepsis, el entorno proinflamatorio provoca en las células mononucleares la expresión de FT en su superficie, que conduce a la activación del sistema de coagulación.63,64
Otra consecuencia de la CID es la inhibición de la fibrinólisis. Además de la disfunción de las células endoteliales durante la sepsis, que también se produce como resultado del ambiente proinflamatorio, el aumento de los niveles del inhibidor del activador del plasminógeno-1 (PAI-1) y del inhibidor de la fibrinolisis activable por trombina (TAFI), dan lugar a alteraciones en la eliminación de la fibrina. Asimismo, el consumo de diversos factores que normalmente regulan la generación de trombina, tales como la antitrombina III, proteína C e inhibidor de la vía del factor tisular (TFPI), contribuye al desarrollo de CID.65,66
La proteína C, la cual es un regulador de la cascada de coagulación, es activada por la trombina unida a trombomodulina y por el receptor endotelial de la proteína C (EPCR) en las células endoteliales. Después de la disociación de EPCR, la proteína C activada se une a su cofactor, la proteína S, que luego resulta en la inactivación de los factores de coagulación Va y VIIIa. Además de su actividad anticoagulante, la proteína C activada tiene propiedades antiapoptóticas y antiinflamatoria profundas. Disminuye notablemente la apoptosis de las células endoteliales y linfocitos y, ejerce efectos pro-fibrinolíticos al inhibir al PAI-1. Los efectos antiinflamatorios de la proteína C activada están mediados a través del EPCR y su unión al receptor activado por la proteasa-1 (PAR1), el cual juega un papel central en la vinculación de la coagulación y la inflamación.67-73
La vía de la proteína C es particularmente susceptible de inhibición como respuesta a la inflamación en la CID mediada por sepsis.61 Además de la disminución en el nivel de la proteína C, la regulación a la baja, la expresión y escisión del complejo trombomodulina-EPCR, son las principales causas de la disfunción de la vía de la proteína C. El HMGB1 inhibe la vía de la proteína C al interferir con el complejo trombina-trombomodulina y también promueve la coagulación por estimulación del FT y la expresión e inhibición del activador del plasminógeno tisular (tPA), una proteasa de serina de la superficie de las células endoteliales, que activa a la plasmina en la cascada de la fibrinolisis.72,73
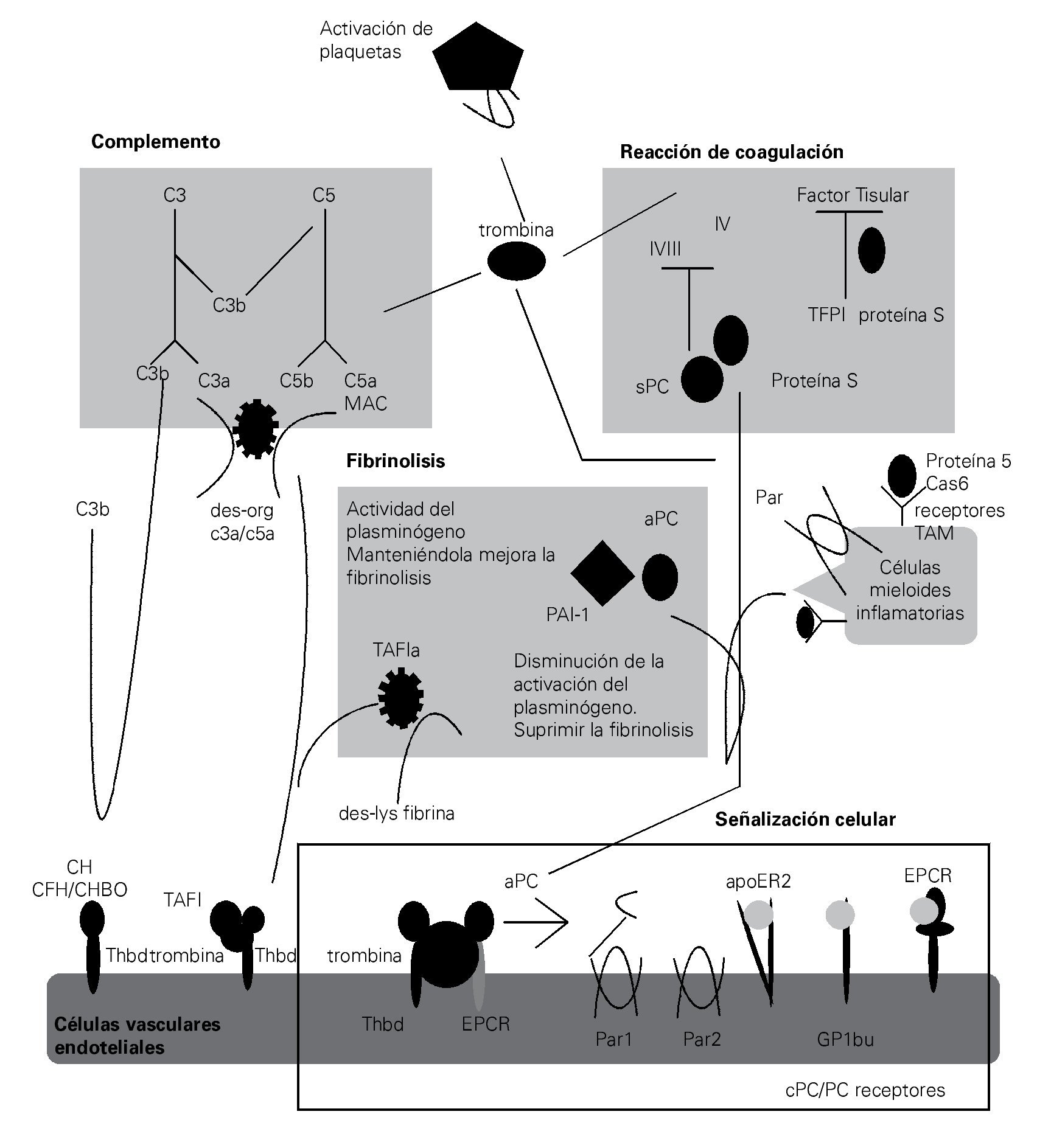
Vinculación entre el sistema del complemento y la coagulación
Tradicionalmente, los sistemas del complemento y de la coagulación se describen como cascadas separadas. Como descendientes de una vía ancestral común, ambas son cascadas proteolíticas compuestas de proteasas de serina con características estructurales comunes y estímulos activadores comunes60 (Figura 2).
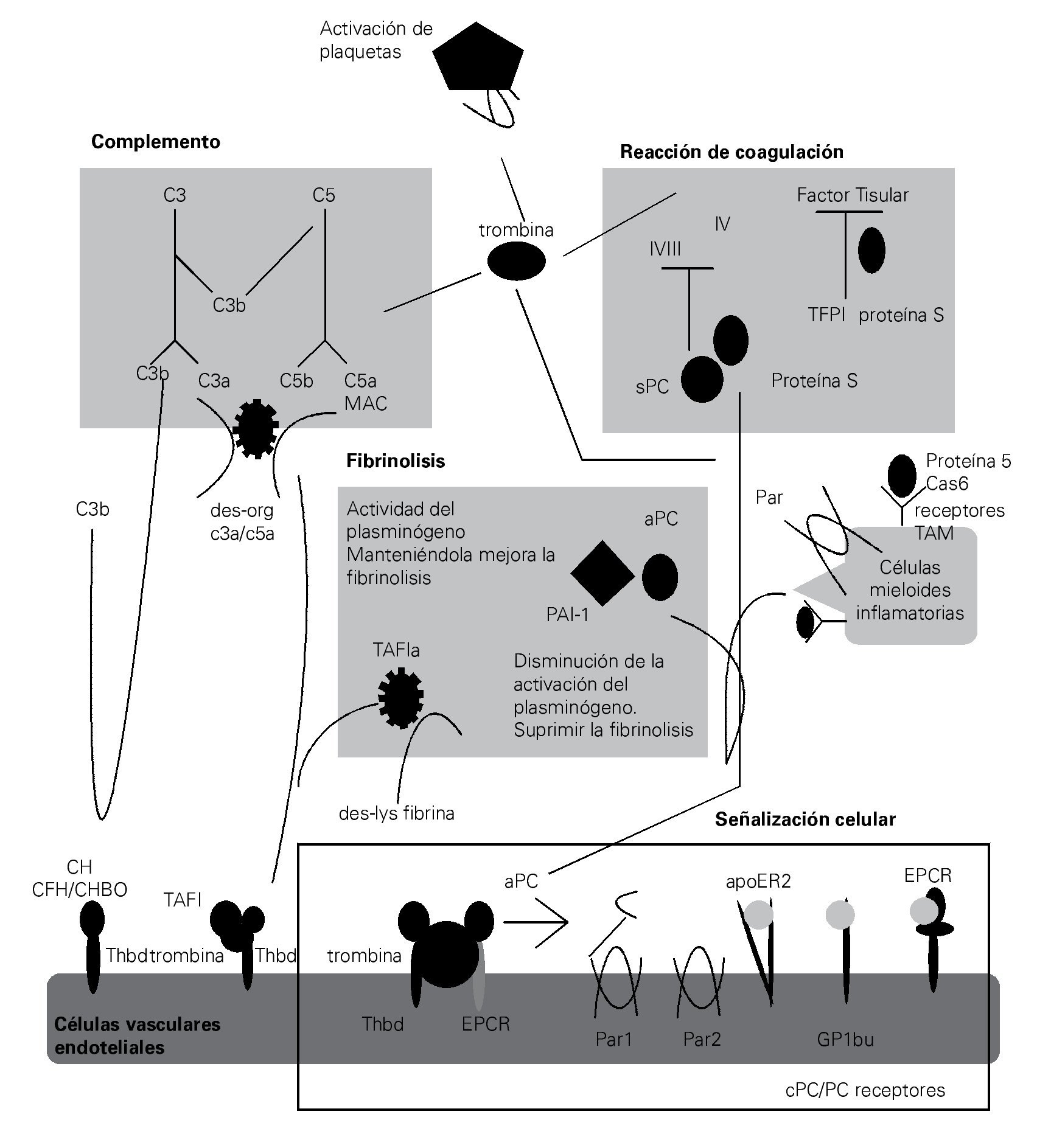
> Figura 2. Esquema en el cual se representa la estrecha relación que existe entre los sistemas del complemento y coagulación en la fisiopatogenia de la sepsis, así como las complicaciones derivadas del imbalance entre ambos sistemas.
Esta relación no se limita a la similitud bioquí-mica de sus proteasas de serina, dado que, estas dos vías también están vinculadas por muchas conexiones mutuas que conforman una red compleja.
Durante la sepsis, la vía de coagulación activada predispone a la trombosis y CID, lo cual puede agravar aún más la respuesta inflamatoria excesiva y activar al complemento. Una interacción bien conocida
entre el complemento y la coagulación es la activación de la vía clásica del complemento a través del factor de la coagulación XIIa, el cual puede activar el componente C1 del complemento.10 De manera más recientemente, se ha demostrado que la trombina puede funcionar como una C5 convertasa en un modo C3-independiente. Esta diafonía es particularmente interesante, no sólo porque la trombina y C5a son factores centrales de sus respectivas cascadas, sino también porque esto indica que C5a y el MAC pueden ser generados en ausencia de la activación del complemento. De forma similar a la trombina, la calicreína y la plasmina se unen directamente C3 y sus fragmentos activos. En un circuito de retroalimentación negativa indirecta, la trombina activada-TAFI inactiva a C3a y C5a.74-76
El sistema del complemento amplifica la coagulación mediante la modificación de los fosfolípidos de las membranas (requeridas para la iniciación de la coagulación a través de FT), por activación de las plaquetas, induciendo la expresión del FT y PAI-1 por los leucocitos.77,78 En consecuencia, el bloqueo de C5a en el modelo experimental de sepsis, mejoró notablemente los efectos de CID.55 Además, la proteasa 2 de serina-lecitina unida a manan (MASP2), una proteasa que es característica en la activación del complemento a través de la vía de la lecitina, puede activar la coagulación mediante la escisión de la trombina en trombina activada.79
La actividad procoagulante del complemento se amplifica cuando los mecanismos anticoagulantes son inhibidos. Además se han documentado múltiples influencias indirectas del sistema del complemento sobre la cascada de la coagulación, que son mediadas a través de otros factores proinflamatorios (TNF, IL-6 y HMGB1).
> Conclusión
La sepsis es un síndrome complejo en el que la activación de la inmunidad innata, induce una intensa respuesta proinflamatoria caracterizada por una intensa respuesta molecular y por un disbalance con los mecanismos reguladores en especial el antiinflamatorio y la coagulación, lo que condiciona disfunción y lesión del endotelio vascular y de la microcirculación.
> Financiamiento
No se recibió ningún patrocinio para realizar este artículo.
> Conflicto de intereses
Los autores declaran no tener conflicto de intereses.
Correspondencia:
Dr. Eduardo E. Montalvo Javé.
Servicio de Cirugía General, Unidad 304,
Hospital General de México "Dr. Eduardo Liceaga".
Dr. Balmis N° 148, Colonia Doctores, Delegación Cuauhtémoc, México D.F., México.
Correo electrónico:montalvoeduardo@hotmail.com.
Dr. Raúl Carrillo Esper,
Unidad de Terapia Intensiva.
Fundación Médica Sur. México D.F.
Correo electrónico:revistacma95@yahoo.com.mx