


Solid pseudopapillary neoplasm of the pancreas is a rare form of cancer with low malignant potential, accounting for 0.17–2.7% of all pancreatic tumours.
Case report 1A 17-year-old female patient who presented with symptoms six months after a Caesarean section. She was referred to our institution for a 10cm×10cm tumour in the upper right quadrant. Abdominal computed tomography showed a mixed mass at the head of the pancreas compatible with Frantz's tumour. Underwent surgery, in which the classic Whipple procedure was performed.
Case report 2A 17-year-old female patient with a history of blunt abdominal trauma days before admission. Abdominal computed tomography showed a 10.6cm×10cm heterogeneous and septate tumour at the head of the pancreas; preoperative diagnosis of solid pseudopapillary neoplasm. Underwent surgery, in which the pylorus-preserving Whipple procedure was performed.
ConclusionSolid pseudopapillary neoplasm of the pancreas is a rare tumour of unknown origin and with good prognosis after surgical resection.
Las neoplasias sólidas pseudopapilares del páncreas son una rara neoplasia de bajo potencial maligno, representan del 0.17 al 2.7% de todos los tumores pancreáticos.
Caso clínico 1Paciente femenino de 17 años de edad, cursando el sexto mes post cesárea. Refieren a nuestra institución por tumoración en cuadrante superior derecho de 10x10cm. Tomografía abdominal reportó una masa con componente mixto dependiente de cabeza de páncreas compatible con tumour de Frantz. Intervenida quirúrgicamente, realizando procedimiento de Whipple clásico.
Caso clínico 2Paciente femenino de 17 años de edad, antecedente de traumatismo contuso abdominal días antes de su ingreso. Tomografía abdominal reporta tumoración heterogénea y septada dependiente de cabeza de páncreas de 10.6x10cm, intervenida quirúrgicamente con diagnóstico preoperatorio de neoplasia sólida pseudopapilar realizando procedimiento de Whipple con preservación pilórica.
ConclusiónLa neoplasia solida pseudopapilar del páncreas es una entidad rara, con un origen desconocido con buen pronóstico con la resección quirúrgica.
Solid pseudopapillary neoplasm of the pancreas was first described by Frantz in 1959 as a rare neoplasm of low malignant potential, accounting for 0.17–2.7% of all pancreatic tumours and 3% of all cystic neoplasms of the pancreas. It is also known as Frantz's tumour, solid and papillary tumours, solid and cystic tumour, papillary cystic neoplasm, and solid papillary epithelial tumour. In 1996 the World Health Organisation defined it as solid pseudopapillary tumour of the pancreas, reclassifying it as solid pseudopapillary neoplasm (SPN), and as low-malignant neoplasm of the exocrine pancreas in 2010.1,2
SPN mostly affects young women in the second or third decade of life, with an average age of 27.2 years, at a female-to-male ratio of up to 9.5:1, while in children the female-to-male ratio is 3:1.3,4
The origin, prognostic factors and natural history of these tumours are unknown, although the tendency to affect young women suggests that sex hormones may be involved in its origin.5
Currently surgical resection is the treatment of choice for SPNs even with local invasion or metastasis. The 5-year overall survival rate is about 95%.1,3
Below we present two cases of SPN in 17-year-old adolescents who were treated at the Hepatopancreatobiliary Surgery and Paediatric Surgery departments of Hospital General de México Dr. Eduardo Liceaga.
Case report 1A 17-year-old female patient who presented with symptoms six months after Caesarean section, with a history of exploratory laparotomy performed 2 months earlier at another hospital, with a preoperative diagnosis of cholecystitis. She was referred to our hospital upon observing pancreatic tumour.
Upon admission, the patient reported a sensation of postprandial fullness, nausea and vomiting, with no other symptoms. Physical examination revealed a good general condition, abdomen with a hypertrophic scar in the infraumbilical midline and a right paramedian scar. A 10cm×10cm tumour was felt on palpation in the upper right quadrant, without signs of peritoneal irritation.
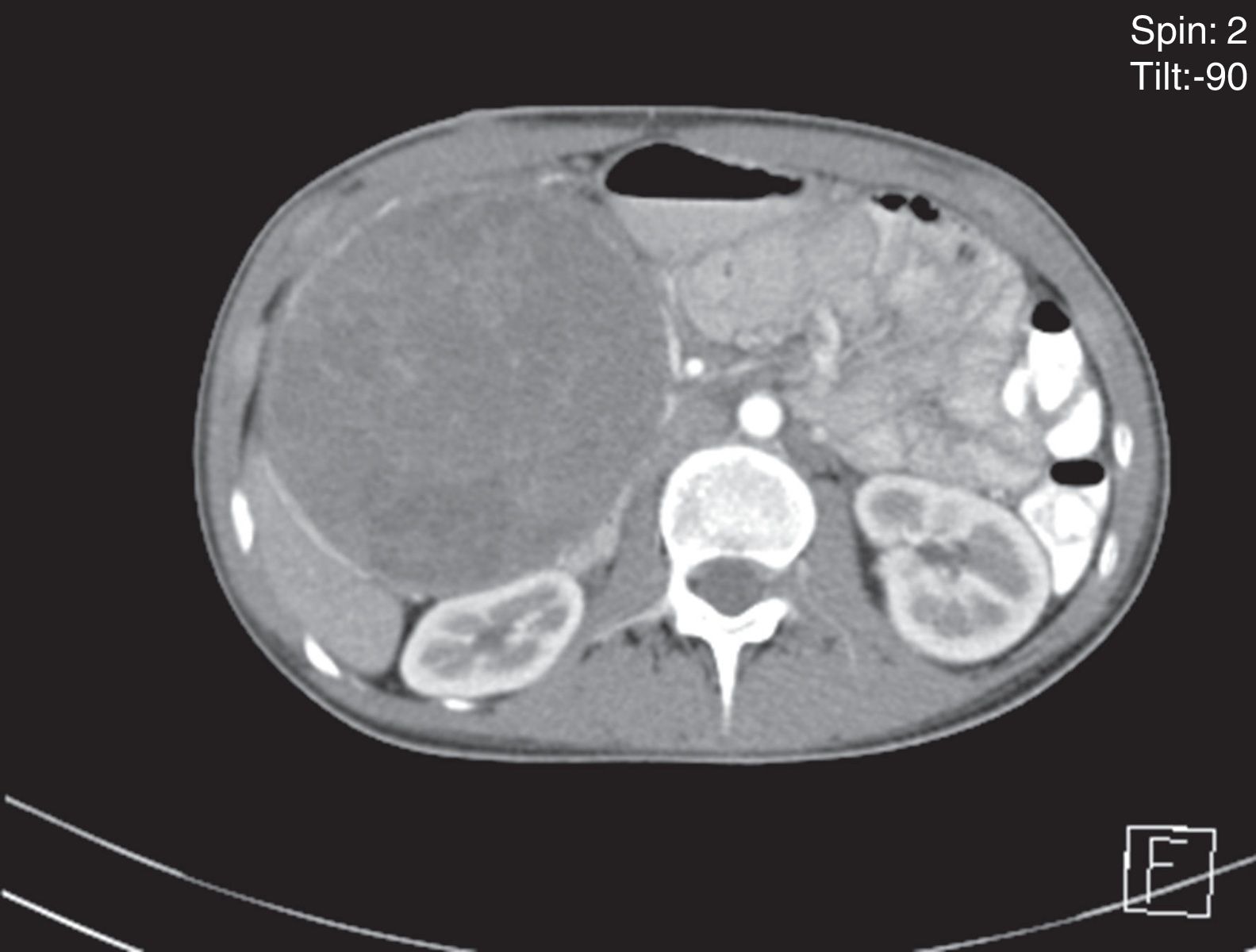
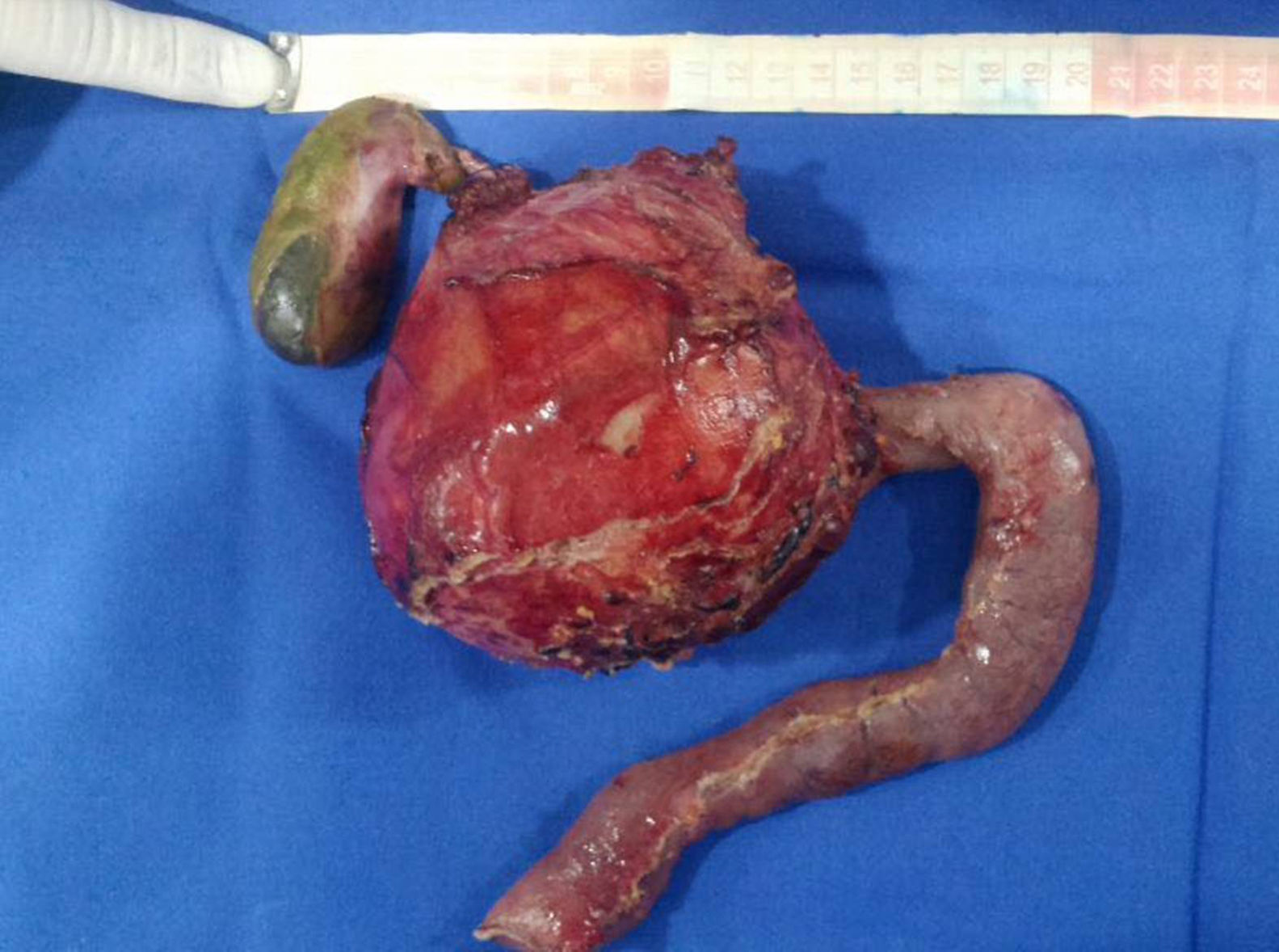
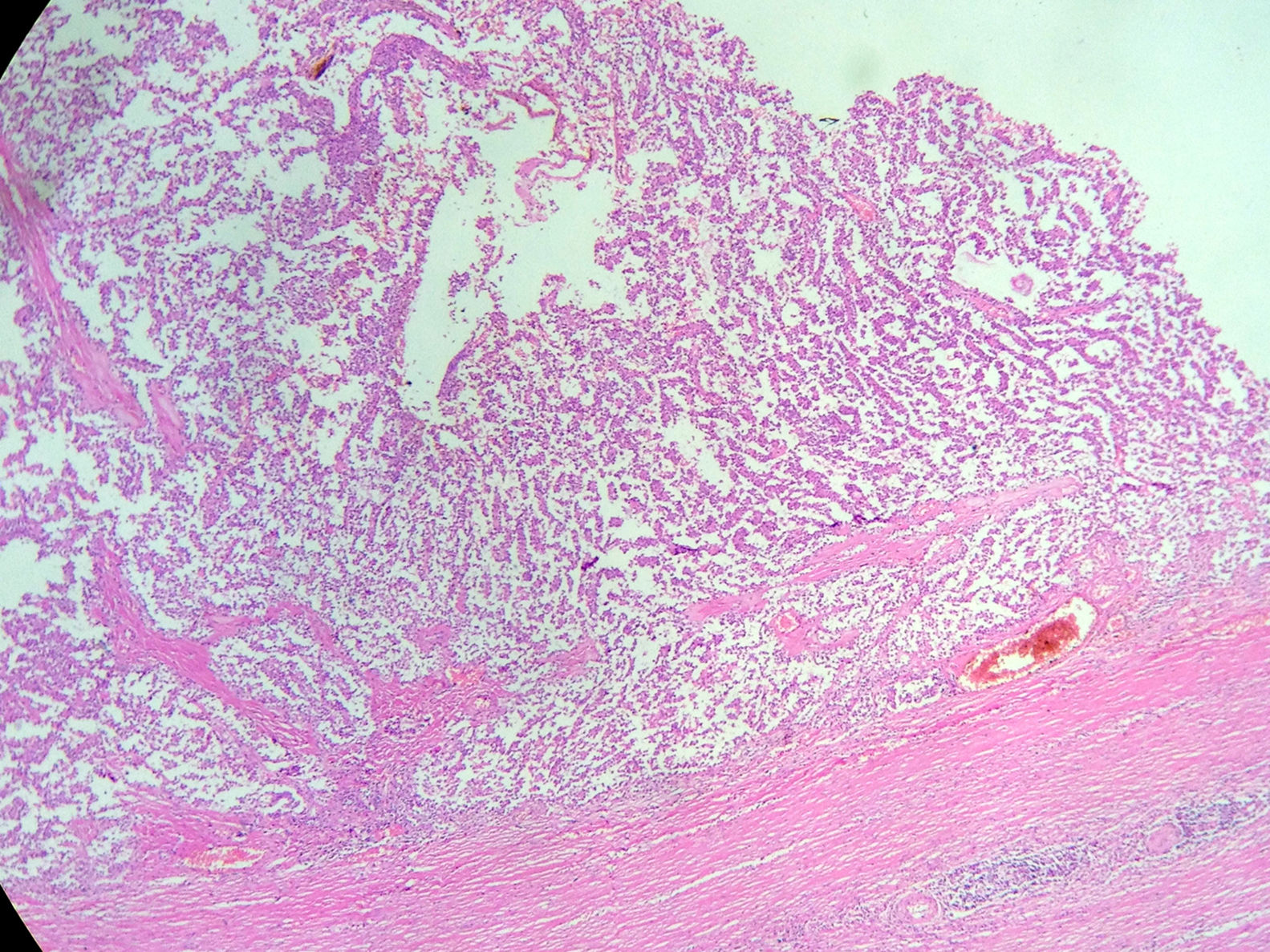
An abdominal ultrasound revealed a round, encapsulated heterogeneous lesion measuring 10.6cm×9.3cm×12.0cm, with a 3.4-mm wall, regular and well-defined margins, a volume of 629.9cc, and no dilatation of the main pancreatic duct. Abdominal computed tomography showed a complex mixed mass at the head of the pancreas that did not result in dilatation of the bile duct or the main pancreatic duct (Fig. 1). The mass was surgically removed by performing the classic Whipple procedure (Fig. 2) with telescope termino-terminal pancreaticojejunal and hepaticojejunal anastomosis measuring 4mm in diameter, and by placing a Penrose drain at the surgical site. The histological study showed a 13cm×10cm×10cm solid pseudopapillary tumour at the head of the pancreas and follicular hyperplasia in 24 lymph nodes, without vascular permeation or invasion of adjacent structures (Fig. 3).
: showing a mixed mass at the head of the pancreas.")
.")

A type “A” pancreatic fistula was documented three days after surgery, which was resolved after 6 days of conservative management. The patient later presented with elevated direct bilirubin levels. Suspecting biliary stenosis because of previous narrow anastomosis, it was decided to place a transanastomotic percutaneous catheter into the bile duct by means of interventional radiology, which corroborated the diagnosis. The patient completed the 17th day of her hospital stay satisfactorily. She is currently on her 12th month of follow-up by the Department of Hepatopancreatobiliary Surgery with a dilatation protocol of the bile duct using a 16Fr percutaneous catheter.
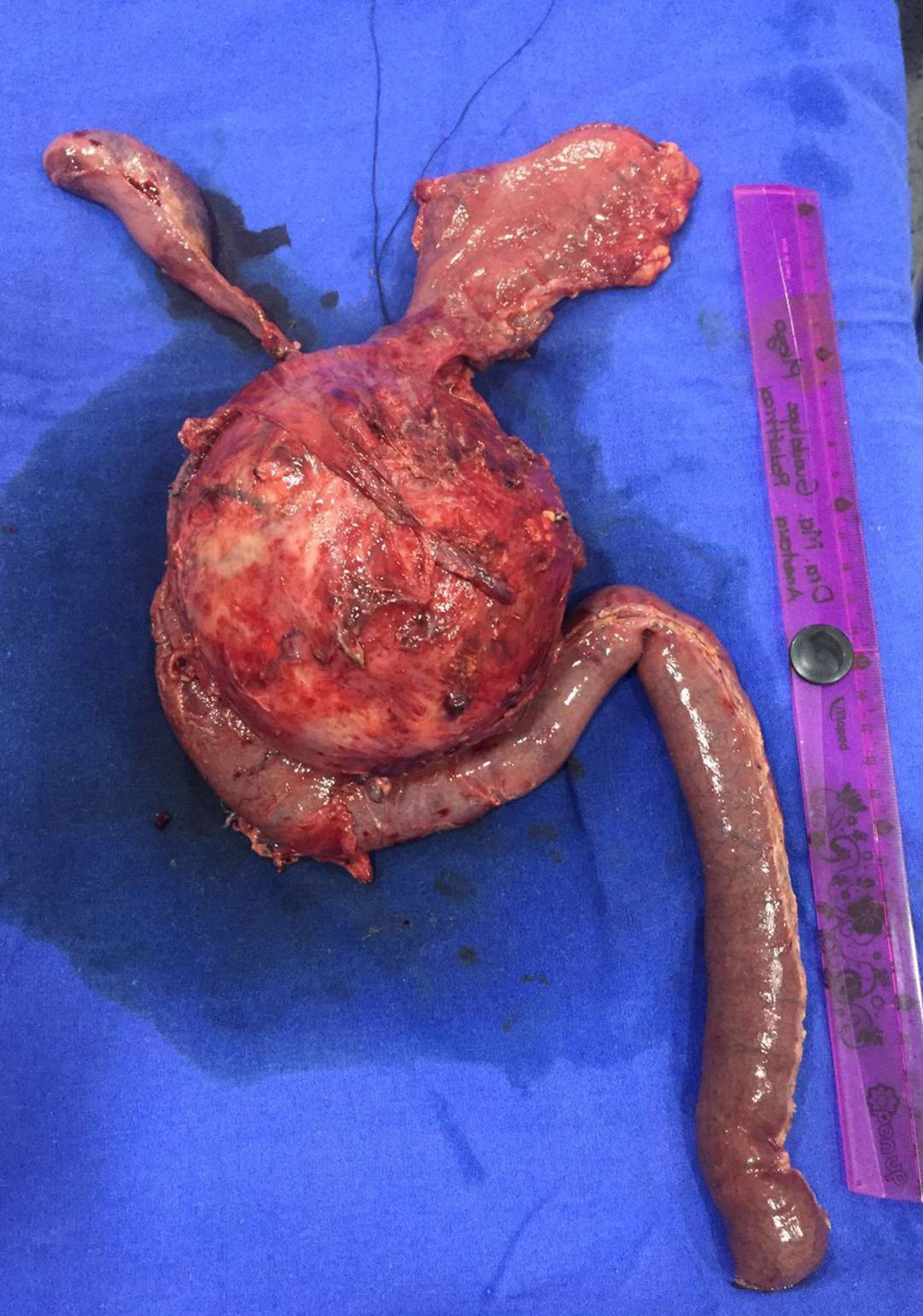
Case report 2A 17-year-old female patient who suffered blunt abdominal trauma days before her admission to hospital. An abdominal ultrasound revealed a 10.4cm×9.5cm×9.5cm heterogeneous, round, septated lesion and calcified areas, with a thickened wall measuring 6mm and a volume of 500.5cc. Abdominal computed tomography showed a 10.6cm×10cm×9.7cm complex mixed mass at the head of the pancreas. Since laboratory tests showed no changes, the patient underwent surgery with a preoperative diagnosis of solid pseudopapillary neoplasm, for which a pylorus-preserving Whipple procedure was performed (Fig. 4). Telescope termino-terminal pancreaticojejunal and hepaticojejunal anastomosis measuring 8mm in diameter was performed, while a Penrose drain was placed at the surgical site. The histological study revealed: solid pseudopapillary neoplasm at the head of the pancreas measuring 12cm×10cm×10cm, with extensive coagulative necrosis, and no tumour at the surgical margins or metastases in 14/14 lymph nodes. The postoperative period was favourable, starting oral administration on day four and discharged on day ten without complications. The patient is currently on her 10th month of follow-up, progressing satisfactorily.
Discussion.")
We searched world and national literature in PubMed, Medline, EBSCO and Medigraphic, using the keywords “pancreatic cyst”, “pancreatic neoplasm”, “solid pseudopapillary neoplasm” and “Frantz's tumour”. A total of 9928 publications were found, reporting 2800 SPN cases in 27 articles with only 5 cases in Mexico. At least 90 cases of SPN in the paediatric population were reported until 1999.4 The number of solid pseudopapillary neoplasms reported increased in the past decade, with a 7-fold increase in the number of cases detected since then.4–9
Because the origin of these tumours has not yet been established, 3 hypotheses have been put forward: (1) They are derived from cells of the pancreatic duct; (2) they originate from acinar cells; or (3) they stem from endocrine cells. Another hypothesis is that SPN arises from pancreatic stem cells or from extra-pancreatic tissue, possibly gonadal (ovaries), which may have been attached to the pancreatic parenchyma during early embryogenesis. Genetic factors have also been linked to the tumour's origin since a higher incidence has been observed in Asian women.1,4
Molecular events are characterised by the presence of the activating mutation of the β-catenin gene that interferes with the phosphorylation of protein products.6 Progesterone receptor expression and strong predilection for women indicate that this could be a hormone-dependent tumour. Sun et al. reported that the hepatitis B virus (HBV) could be involved in the pathogenesis of SPN; however, this association has not been confirmed by other researchers.1,3,10
Clinical presentation is non-specific, with most patients presenting with unclear clinical characteristics including mild abdominal pain, hyporexia, nausea, vomiting, weight loss and, very rarely, jaundice, pancreatitis and haematemesis (in less than 12%)—and a third of patients are asymptomatic. Eight per cent (8%) of the patients present with acute abdomen as a result of blunt abdominal trauma or spontaneous rupture of the capsule. Fortunately, the young woman in our second case did not develop acute abdomen after the trauma.1–3,9
Due to their slow growth and asymptomatic presentation, as in the second reported case, tumours are large in size by the time of diagnosis. The average size is 9.5cm, but tumours up to 30cm have been reported. In our reported cases, the tumours measured 13cm and 12cm respectively. The tumours are mostly found in the body of the pancreas, followed by the tail. Nevertheless, some case series report a greater number of tumours located at the head of the pancreas (39.8%), as in the two cases presented in this article.1,11
Macroscopically, SPNs have cystic and solid components with haemorrhagic areas, fibrous capsules and, at times, calcifications. The key histological characteristic is the solid and pseudopapillary proliferation of homomorphic cells without increased mitosis or cytologic atypia. Immunohistochemically, they include positive staining for β-catenin, vimentin, α1-antitrypsin, α1-chymotrypsin, neuron-specific enolase, cyclin D1 and, more recently, negative E-cadherin membrane. Positivity for the progesterone and oestrogen receptors varies, with more positivity for the oestrogen receptor. They may also reveal focal immunoreactivity for cytokeratin and synaptophysin and may express galectin-3, all of which are useful in differentiating SPN from pancreatic endocrine tumours.1,3,12
The Ki-67 index has been suggested as a possible marker for malignant potential; a low value on the index (≤5%) indicates slow tumour growth.13
Definitive histopathological criteria for malignancy are not well established. However, a tumour larger than 5cm, with a high proliferation rate, pancreatic parenchymal infiltration, vascular/lymphatic/perineural invasion and invasion of adjacent structures, as well as nuclear atypia and distant metastasis, can predict aggressive behaviour and be designated as solid pseudopapillary carcinoma.11,13 Preoperative diagnosis of SPN is difficult since patients lack distinctive symptoms, and so most of these tumours are diagnosed during complementary imaging tests.3,13 Ultrasound, abdominal computed tomography and magnetic resonance usually show a markedly well-circumscribed heterogeneous mass, with different solid and cystic components, usually delimited by a peripheral capsule and occasional calcifications, similar to our two cases. Positron emission tomography is not used because it may not provide additional information.11 Intratumoural haemorrhage and septa are characteristic of SPN. A preoperative histological diagnosis can be made by endoscopic ultrasound (EUS), fine-needle aspiration (FNA), or ultrasound- or CT-guided percutaneous core needle biopsy.1,14 The use of ultrasound-guided percutaneous or endoscopic fine-needle aspiration cytology (FNAC) may help to distinguish SPN from other pancreatic tumours when a radiological diagnosis is not clear enough.3
To date, radical surgical resection is the treatment of choice for SPNs, even with local invasion or metastasis. Extensive lymphatic dissection or resection of adjacent structures is not justified because of the rare incidence of metastasis in lymph nodes (less than 2%).1,3,11
Although the malignant potential of SPN is low, approximately 10–15% of patients develop metastatic disease, which often involves the liver or peritoneum. Therefore, all patients need long-term follow-up.1 To date, the role of chemotherapy and radiotherapy in the treatment of SPN is poorly defined, with few reports available so far.10
ConclusionSPN of the pancreas is extremely rare and is usually detected incidentally. Its natural origin, prognostic factors and natural history are unknown, and it has an excellent prognosis after surgical resection.
Ethical disclosuresProtection of human and animal subjectsThe authors declare that no experiments were performed on humans or animals for this study.
Confidentiality of dataThe authors declare that they have followed the protocols of their work centre on the publication of patient data.
Right to privacy and informed consentThe authors have obtained the written informed consent of the patients or subjects mentioned in the article. The corresponding author is in possession of this document.
Conflict of interestThe authors declare that they have no conflicts of interest.