


Aggressive angiomyxoma is a rare type of soft tissue tumour. It has traditionally been described as a low-grade myxomatous neoplasm of the vulvovaginal, perineal and pelvic regions in women, and has a high rate of recurrence because of its infiltrative potential and locoregional extension. Its pathogenesis and histogenesis are poorly understood, but it is proposed that chromosome 12 alterations and fibroblastic/myofibroblastic tumours are involved in its origin. Around 250 cases have been reported to date in the western literature, some of them with unusual presentations. The biggest reported tumour measures 26cm×21cm×6cm. The following article describes two case reports of this neoplasm, which were studied at Mexico's General Hospital “Dr. Eduardo Liceaga” over period of one year. Both case reports state that the tumours are considerably bigger than the ones currently reported (66cm×60cm×20cm and 50cm×46cm×17cm). We shall focus on histopathology and end with an analysis and discussion.
El angiomixoma profundo o agresivo es una entidad poco frecuente, agrupada dentro de las neoplasias del estroma genital. Se describe clásicamente como una neoplasia mixomatosa de bajo grado de las regiones vulvovaginal, perineal y pélvica en mujeres, con una tasa de recurrencia alta debido a su potencial de infiltración y extensión locoregional; con patogénesis e histogénesis pobremente entendidas, postulándose alteraciones en el cromosoma 12 y origen fibroblástico/miofibroblástico. La literatura occidental reporta poco más de 250 casos a la fecha, algunos de ellos con presentaciones inusuales, el caso informado de mayor tamaño es de 26×21×6cm. A continuación se describen dos casos de dicha neoplasia estudiados en el Hospital General de México “Dr. Eduardo Liceaga”, en el periodo de un año, ambos de tamaños considerablemente mayores a los reportados actualmente (66×60×20cm y 50×46×17cm) y se hace enfoque en su estudio histopatológico, finalizando con un análisis y discusión sobre el tema.
Deep or aggressive angiomyxoma (AA) is a rare condition, grouped within the genital stromal tumours, which include stromal fibroepithelial polyps, superficial cervico-vaginal myofibroblastoma, cellular angiofibroma, myofibroblastoma of the breast, angiomyofibroblastoma and aggressive angiomyxoma. They are included in this group because they originate in the mesenchymal subepithelial region, which encompasses the endocervix of the vulva, with immunophenotypic and genetic similarities.1,2
It has traditionally been described as a low-grade myxomatous neoplasm of the vulvovaginal, perineal and pelvic regions in women.1,3
Though it is considered a low-grade tumour, it has a high rate of recurrence because of its infiltrative potential and locoregional extension.1–5
Its pathogenesis is poorly understood, but some cases have found chromosome 12 alterations, mainly in the 12q13-15 region,3,4 in relation to the high mobility group I-C gene (HMGI-C).3
There is a lack of consensus regarding its histogenesis, with the most likely source being fibroblasts/myofibroblasts and in the primitive multipotent mesenchymal cells in the lower genital tract, and it can be differentiated in various ways.
This condition was first described by Steeper and Rosai in 1983, and at the time this review was conducted, the western literature had reported approximately 250 cases. In addition, cases with unusual presentations were reviewed: a male patient with presacral space involvement,6 presentation of an abdominal tumour, occupying the path of the inferior vena cava and right atrium7 and an abdominal tumour extending to the perineum and herniation into the gluteal region8; the largest size reported was 26cm×21cm×6cm.3
Case reportsHere we describe two cases related to this condition occurring at Hospital General de México between June 2014 and April 2015. In each case, multiple sections were performed, stained using the haematoxylin-eosin technique. Furthermore, additional paraffin-embedded sections were selected, using antibodies against smooth muscle actin, desmin, vimentin, oestrogen receptor, progesterone receptor, b-catenin, S100 and CD34 and electron microscopy was performed in both cases.
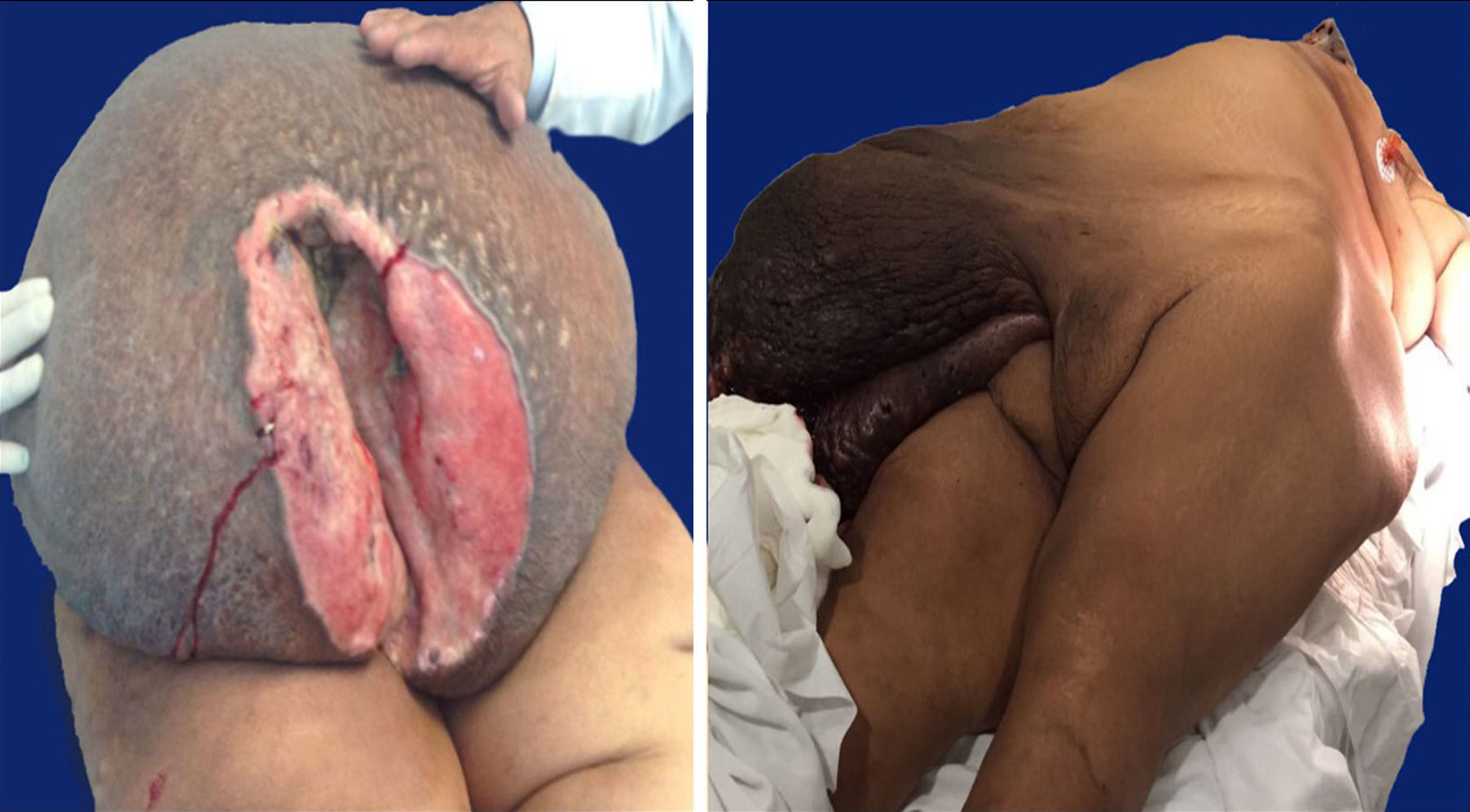
Case 127-year-old woman with a family history of type 2 diabetes mellitus on the sides of both parents; her father had lung cancer (unspecified), and she has a personal medical history of grade 3 obesity (BMI 56.3); no other relevant history. For 6 years she experienced symptoms characterised by a progressive increase in inguinal-region volume, extending to all the lower half of the abdomen, with dyspnoea on moderate effort during the prior year and ulceration of the area with 8 months of history, after which time she went see the doctor. There was a pendulous mass of about 25kg with infiltration of the skin covering it, extending from the inguinal region to mesogastrium, with hyperpigmentation and a significant collateral venous network in the legs due to an obstructive event (Fig. 1). The examination revealed no other changes. An ultrasound showed a tumour on the abdominal wall, with no other features. She underwent an oncologic resection with reconstruction, with a soft-tissue tumour on the anterior abdominal wall, with a depth reaching to fascial plane, in addition to an umbilical hernia with the omentum inside. The histopathological report stated that it was an aggressive angiomyxoma. Her postoperative period was without complications. She was assessed by the Radio-oncology Department, which decided she needed radiation therapy. However, this was not feasible due to the patient's morbid obesity, and so she was sent to the Endocrinology Department for consultation. There are no recurrence data from the follow-up visits during the year following the surgery.
Case 2.")
40-year-old woman with a history of hypertension, diagnosed 10 years ago and controlled with captopril, hypothyroidism, diagnosed 1 year ago, with no treatment specified, and grade 3 obesity (BMI 50.8). For 8 years she had symptoms characterised by a progressive and painless increase in volume in inguinal region on her right side, extending to the lower half of the abdomen, predominantly on the right side, which caused her trouble walking due to the tumour volume during the 3 months prior to her admission. She was seen at Hospital General de México, where a pendulous mass originating from the right labia majora was found, extending to the lower half of the abdomen, with an ovoid ulcerated area 4cm in diameter and 7cm deep, with a clumpy greyish red substance (Fig. 1). An abdominal/pelvic CT scan showed a soft-tissue tumour on both sides of the anterolateral abdominal wall, as well as an uncomplicated umbilical hernia. She was examined and underwent surgical resection with reconstruction. She had a stable postoperative period, with an uncomplicated infection of the surgical incision, which resolved with no complications. She was followed up by the Oncology Department. An MRI was performed 4 months after surgery, and it showed no signs of recurrence. At her follow-up 7 months after surgery she reported no symptoms, with minimal wound dehiscence and no other abnormalities.
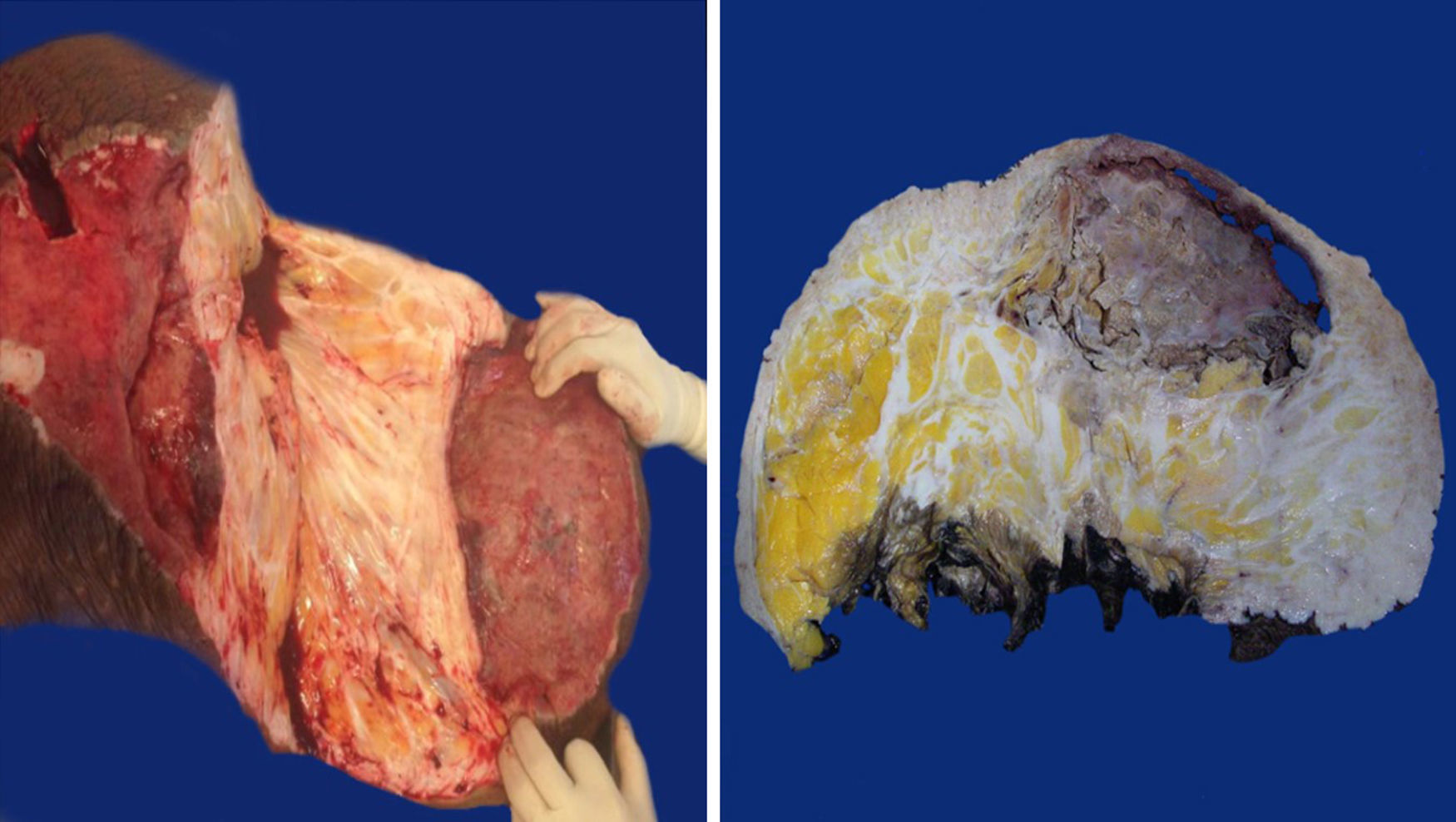
Macroscopic findingsBoth tumours were nodular-shaped and covered by skin. The tumour in the first case measured 66cm×60cm×20cm and weighed 35kg, and the second measured 50cm×46cm×17cm and weighed 16kg. Both had skin ulcers. The first tumour measured 25cm×25cm, and the second 6cm×5.5cm. Both had irregular edges, thickened with crumbly, friable substances in them. They had heterogeneous section surface area, dominated by poorly defined areas with a myxoid, light yellow appearance, separated by multiple irregular fibrous bands that were light grey and rubbery in consistency, as well as areas of opaque yellow soft fat. In both cases the surgical site shows lesions. Furthermore, the second tumour showed a skin ulcer with a connection to a central abscessed area of 10cm×8cm, located in the middle third of the tumour 7cm under the skin (Fig. 2).
Microscopic findings.")
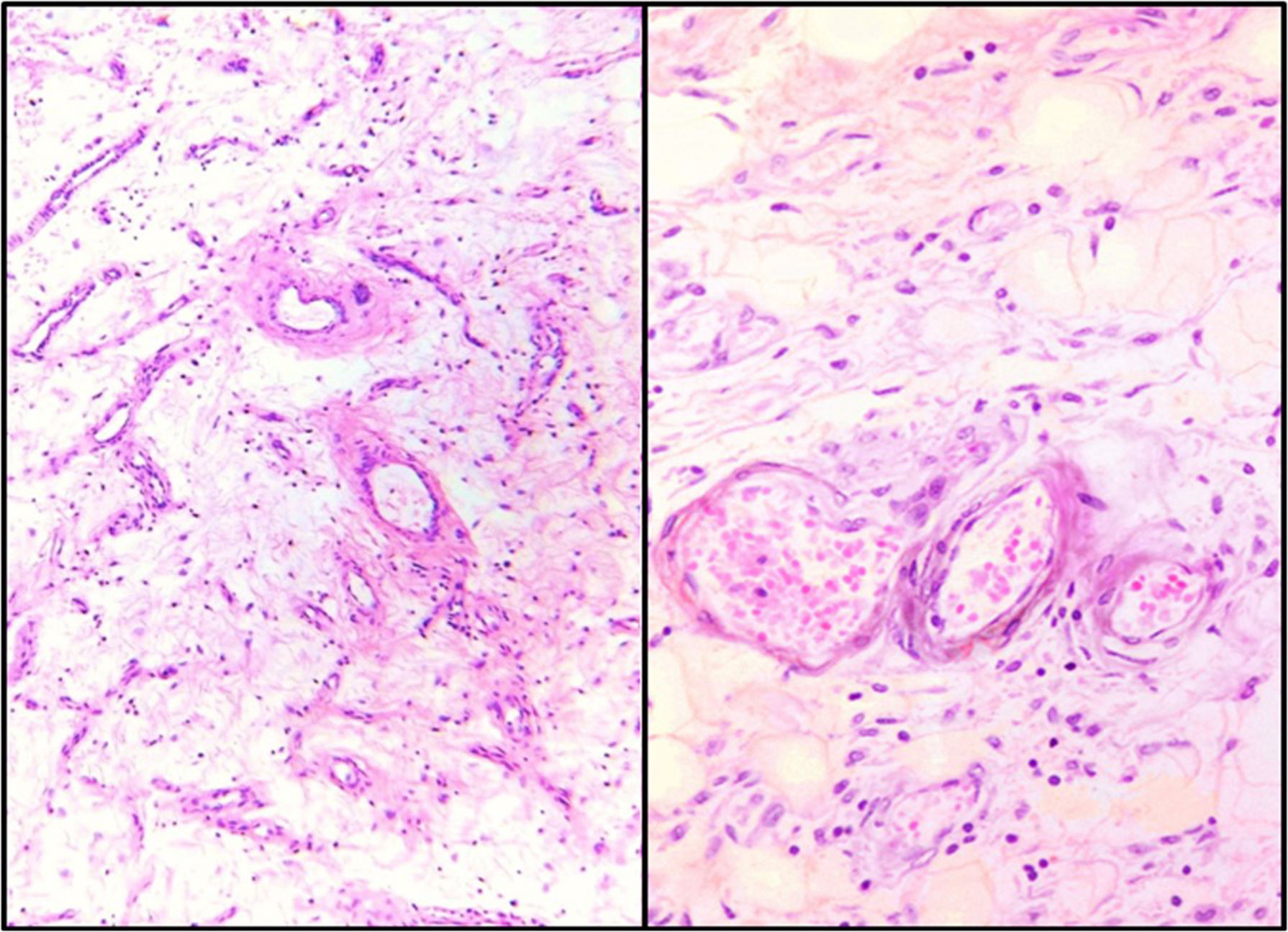
Both showed similar characteristics with light microscopy: a hypocellular lesion with scattered cells with a myxoid background, fusocellular-shaped and some stellate cells, with ill-defined clear eosinophilic cytoplasm, ovoid nuclei with soft chromatin and with no atypia or mitotic activity. Heterogeneously distributed over the lesion, small and medium-sized vessels were also observed with a thickened wall with hyalinisation and concentric hypertrophy of the medial wall, around which scattered neoplastic cells were seen, appearing to “fray” from their walls. In addition, adipose tissue was seen, trapped by the lesion, which extends from the superficial dermis to the surgical site in both cases (Fig. 3).
Immunohistochemistry.")
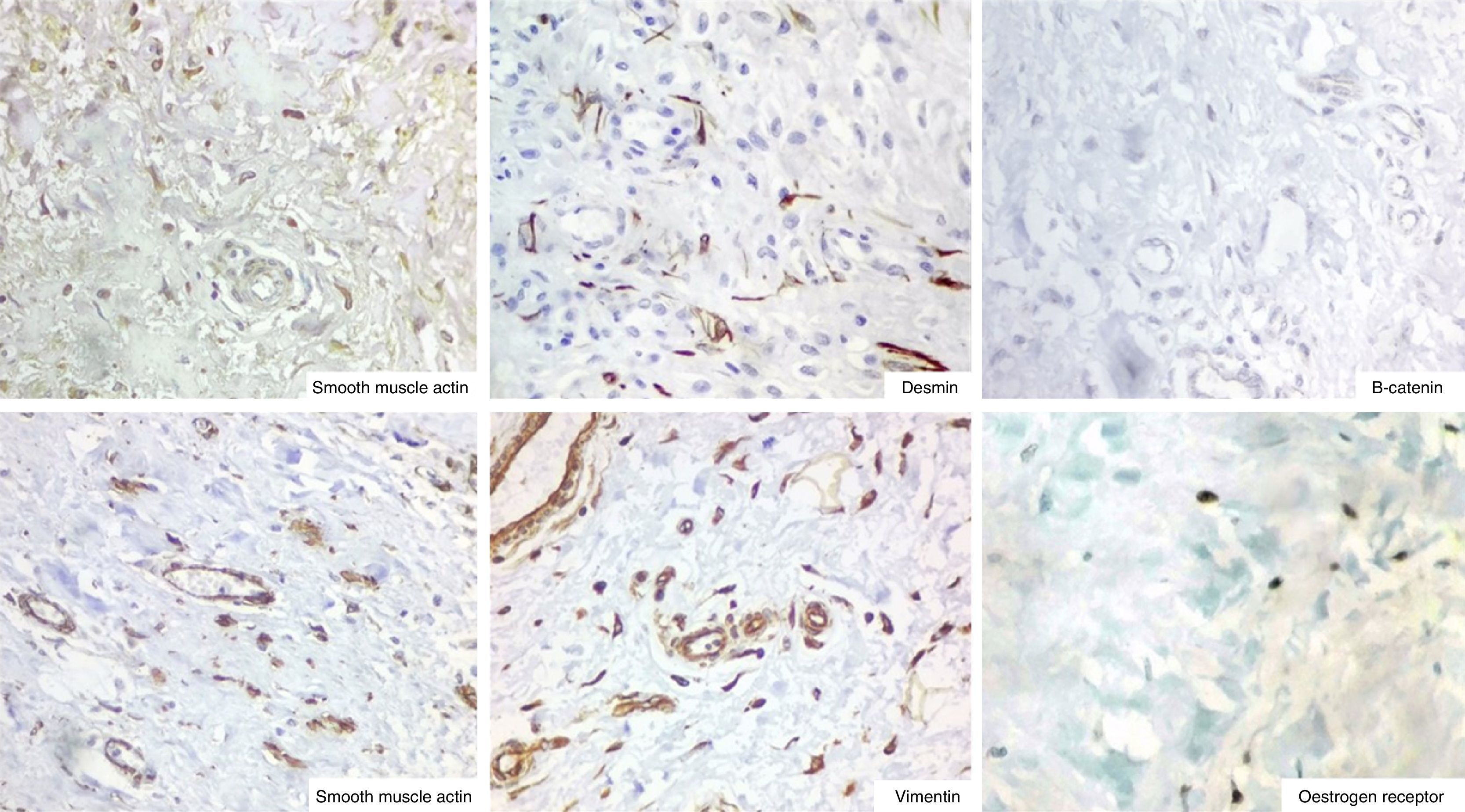
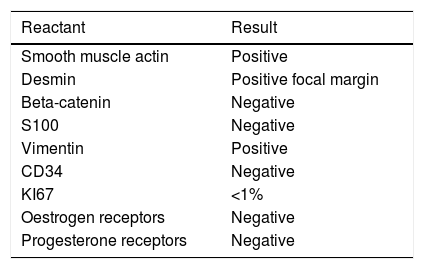
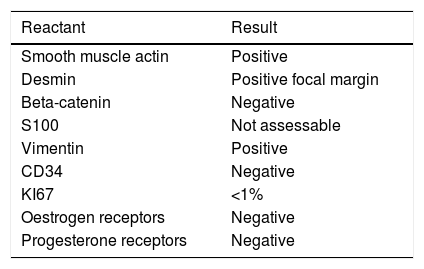
The immunohistochemistry panel below was performed on representative fragments from each case, with the following results (Fig. 4; see Tables 1 and 2).
.")
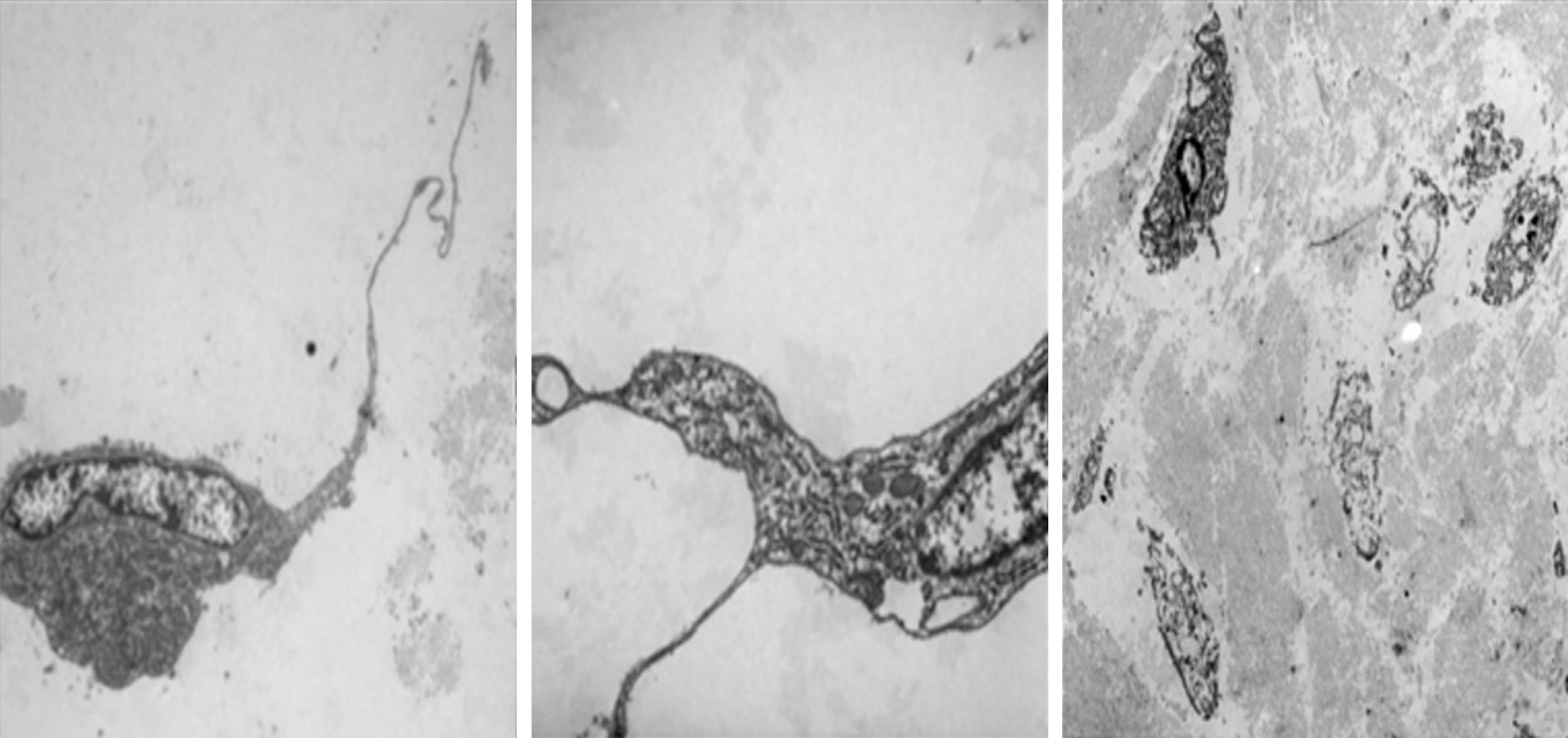
Electron microscopy was performed on the two sections, which showed fibroblastic and myofibroblastic differentiation in the tumour cells; this was useful in order to rule out neoplasms originating in adipose tissue (Fig. 5).
Discussion.")
As in these two cases, the condition predominantly affects women at a ratio of 6.6–1, in the fourth decade of life (although it has been reported at both ends of life).1–4
Regarding imaging, the main method reported for assessment is the use of CT scans, which show tumours with circumscribed edges with less attenuation than with the muscle planes in relation to the myxoid stroma and high water content, alternating with irregular, swirl-shaped areas of increased intensity inside. Regarding MRI, T2 hyperintense and T1 isointense signals are shown. Imaging studies are important for pre-surgical evaluation and for post-surgical follow-up, because the extent of the tumour can easily be underestimated in a physical exam, and because early recurrences can be detected.9
Macroscopically, polypoids are usually greater than 10cm, with the largest case found in the literature to be 26cm×21cm×6cm.3 The surface section appears partially encapsulated and fibrogelatinous in appearance, with poorly circumscribed edges intermingled with adjacent tissues.1,3,5
Microscopically, this is a low to moderately cellular lesion, composed of fusiform to stellate cells with poorly defined cytoplasm and small nuclei that are round to oval, hyperchromatic, with no cytologic atypia or mitotic activity, embedded in a myxoid background with many mucopolysaccharides that often have foci with lots of collagen and bleeding. Among these, there is a variable number of small to medium-sized vessels, with a thickened wall secondary to medial hypertrophy and vascular and perivascular hyalinisation. With regard to the edges of the tumour, there is an interface of infiltration, mixed with non-cancerous adjacent tissues. Light microscopy is considered the most important criterion for diagnosis. All these characteristics were present in both cases here.1–5,9
In immunohistochemical terms, in the different cases reported, the fusiform/stellate cells show varying positivity for actin, desmin, smooth muscle actin, and oestrogen and progesterone receptors, including the cases reported in men. They also show constant negativity for carcinoembryonic antigen, cytokeratin and S100.2–4
Regarding the differential diagnosis, it is important to recognise that most genital stromal tumours, as well as many smooth muscle tumours, may present areas with myxoid foci, and it is therefore important to consider the different differential data in the diagnosis. Smooth muscle tumours, even in their most myxoid forms, are more cellular than is aggressive angiomyxoma, and they present classic areas of intersecting fascicles. What's more, cytologically they are composed of fusiform cells with eosinophilic cytoplasm and nuclei with blunt ends. Well-differentiated liposarcoma falls within the differential diagnosis, but the presence of cytologic atypia rules out the diagnosis of aggressive angiomyxoma. This underscores the importance of taking good samples of the lesion. Also, MDM2 gene amplification supports the diagnosis of liposarcoma. Another variant of liposarcoma, i.e. myxoid, can be misleading; however, the plexiform capillary network and ovoid cells (rather than fusiform cells) help to differentiate it as well as the single translocation of the condition, t(12; 16) (q13; p11). Surface angiomyxoma tends to be smaller and is lobe-shaped, and as its name implies, it is located in the dermis or subcutaneous tissue.1,10
The treatment of choice involves complete excision with close long-term monitoring. Adjuvant hormone therapy has also been suggested, with gonadotropin-releasing hormone analogues as the only ones that have shown efficacy in reducing tumour size; other hormonal and chemotherapeutic agents have not shown efficacy. Radiation therapy plays nothing more than a small role in adjuvant treatment for this condition, because of the low mitotic index.
Regarding prognosis, it is important to recognise this condition, because it is the only tumour of the genital stroma that exhibits a significant risk of recurrence, especially due to its infiltrative edges, which are not always clinically or surgically evident, resulting in reported recurrence rates from 10% to more than 70% up to 7 years after resection, depending on the invasiveness of the surgical procedure.
ConclusionsDeep or aggressive angiomyxoma is a neoplasm with a low incidence rate, in which histopathological examination is the gold standard for diagnosis. The local infiltrative behaviour and high rates of local recurrence involved are important to recognise, because, in the differential diagnosis of this disease, there are multiple benign conditions, as well as multiple sarcomas of varying grades.
In this review the size in both cases stands out as the largest reported to date in the literature in English (26cm×21cm×6cm), as does the frequency of both tumours, considering the number of cases reported to date since the first report.
Ethical disclosureProtection of human and animal subjectsThe authors declare that no experiments were performed on humans or animals for this study.
Confidentiality of dataThe authors declare that they have followed the protocols of their work centre on the publication of patient data.
Right to privacy and informed consentThe authors declare that no patient data appear in this article.
Conflicts of interestThere are no conflicts of interest in the writing or the publication of this document.