


Las malformaciones craneofaciales aisladas o sindromáticas representan un problema de salud pública, debido a las comorbilidades que originan e incapacidad a la que predisponen. Los avances en biología molecular han permitido dilucidar los genes involucrados en múltiples malformaciones. Asimismo, la cirugía reconstructiva y la rehabilitación representan un área elemental en el manejo de estos pacientes. Presentamos a continuación una revisión sobre las bases moleculares, técnicas de diagnóstico molecular, imagenológico y clínico, así como una breve reseña sobre el tratamiento quirúrgico.
Craniofacial malformations, syndromatic and non- syndromatic represent a public health issue because of the comorbidities associated and the incapacity which they predispose to. Novel techniques in molecular biology are aiding to dilucidate the genes involved in several of these malformations. Furthermore, both reconstructive surgery and rehabilitation represent an elemental area in the management of these patients. We present a review on the molecular basis, molecular, imagenological and clinic diagnosis techniques, as well as a brief review on current surgical treatment.
¿ Reseña histórica
Fue Hipócrates quien realizó la primera descripción de malformaciones craneales, asociándolas a la sutura involucrada. El médico particular de Juliano El Apóstata, llamado Oribasio de Pérgamo reportó malformaciones craneofaciales, asociadas a deformidades palatinas.1 Hacia 1800, el alemán Samuel Sommerring describió el crecimiento y función de las suturas craneales, treinta años después, Otto observó que el cierre prematuro de las suturas, impedía el crecimiento apropiado de cráneo compensando éste, a expensas de las suturas no afectadas.2 Posteriormente, éstas observaciones condujeron a Virchow3a la introducción del término craneosinostosis, definido como malformación caracterizada por la fusión prematura de una o más suturas craneales. A partir de ese momento, han sido varios los médicos que han asociados a las craneosinostosis con otras alteraciones. Fue Thomson, quien realizó estudios de audición en sujetos con malformaciones de oído externo en 1846.4 Berry describió colobomas en el párpado inferior en 1889,5 mientras que en una reunión de la Sociedad Londinense de Oftalmología en 1900, Treacher Collins presentó a dos pacientes con malformaciones oculares y defectos de los huesos malares.6 Finalmente en 1944, Franceschetti, Zwahlen y Klein publicaron una extensa revisión de las manifestaciones clínicas en sujetos, con las malformaciones previamente descritas por Collins, asociándolas a un síndrome, al que llamaron la disostosis mandíbulo facial o Síndrome de Treacher Collins. Mientras que en diversas regiones europeas, es conocido como el Síndrome de Franceschetti-Klein.7-9 Durante el siglo XX se realizaron avances en el manejo de estos pacientes, se realizaron las primeras clasificaciones incluyendo a las malformaciones de labio y paladar.10-12
Durante las últimas dos décadas del siglo XX y actualmente en el siglo XXI, las grandes y novedosas técnicas de biología molecular nos han brindado la oportunidad de conocer la complejidad de la morfogénesis craneofacial, identificando el papel de varios genes implicados en el desarrollo óseo, como son por ejemplo, los genes de la familia HOX, SHH, los factores de crecimiento fibroblástico, los retinoides, las proteínas morfogénicas del hueso, los genes involucrados en la craneosinostosis y las metaloproteinasas de matriz. Actualmente, existe la posibilidad de realizar diagnósticos oportunos, abordajes multidisciplinarios adecuados y mejor conocimiento de fisiopatología de éstas enfermedades.
¿ Bases moleculares
El desarrollo craneofacial comprende un complejo proceso caracterizado por proliferación, diferenciación y migración celular de las células neurales. Éstas migran a diferentes regiones del embrión, donde se diferencian en múltiples tipos celulares como neuronas periféricas, neuronas entéricas, células gliales, melanocitos, músculo liso y condrocitos. En la actualidad, gracias a los avances en biología molecular y genética médica, conocemos el papel fundamental de múltiples moléculas, que participan en la morfogénesis craneofacial.
La formación de las estructuras craneofaciales inicia con la formación de las células de la cresta neural, para ello se requiere de la participación de proteínas antagónicas de la proteína morfogenética de hueso (BMP) y factores de crecimiento de fibroblastos, entre otros. Posteriormente, en el dorso del tubo neural ocurre señalización mediada por contacto entre los tejidos, ocasionando que las células del neuroectodermo y no neuroectodérmicas de los bordes, pasen por una transición de células epiteliales a mesenquimales, altamente invasivas. Para que las células de la cresta neural migren desde el dorso del tubo neural hasta la región craneofacial, se requiere que pierdan su polaridad ápico basal y simultáneaápico basal y simultáneapico basal y simultáneamente, se liberen complejos de adhesión intercelular quimiotácticos como FGF-2 y FGF-8. Una vez que han llegado a su destino, proliferan para formar las estructuras craneofaciales. Múltiples factores inducen y regulan esta proliferación, como las proteínas Sonic Hedgehog SHH (erizo sónico), el cual se expresa en el ectodermo de los procesos frontonasal y maxilares, el neuroectodermo y el endodermo faríngeo, en diversas etapas del desarrollo y el gen TCOF1.13
Una vez que las células de la cresta neural ocupan los arcos faríngeos, también tienen en su centro una masa de células derivadas del mesodermo rodeadas por CCN, cubiertas en el exterior por ectodermo y en el interior por endodermo. Las interacciones entre estos componentes tienen múltiples efectos críticos en el desarrollo craneofacial.
Diversos estudios demuestran que la ausencia de SHH, origina malformaciones como la holoprosencefalia, hipotelorismo, ciclopia, hipoplasia medio facial, así como labio y paladar hendido.14-16 Los genes Hox participan en el desarrollo axial del embrión, se expresan en la cresta neural antes y después de la migración a los arcos branquiales, anomalías en la expresión resultan en la malformación de cualquiera de las estructuras derivada de los arcos.14,16 En cuanto a las craneosinostosis sindrómicas, se han identificado mutaciones relacionadas en genes de la familia de los receptores de factores de crecimiento fibroblástico (FGFRs).17-19 Mutaciones en el gen FGFR1 se han relacionado con el Síndrome de Pffeifer,18 mutaciones en el gen FGRF2,1 se han asociado a síndromes de Pffeifer, Crouzon, Apert, Jackson-Weiss y Beare Stevenson.17,18,20-24 Mutaciones en el gen TWIST se relacionan al Síndrome de Saethre-Chotzen.25-27 Por otra parte, el gen TGFb3, miembro de la familia de factores de transformación y crecimiento, ha demostrado ser requerido para la fusión palatina.28
Los estudios en moléculas teratogénicas han mostrado los efectos que se pueden tener sobre el genoma, un ejemplo que ilustra lo anterior es el ácido retinoico. Aunque su papel como teratógeno es conocido desde hace 60 años, es hasta hace algunos años que se confirmó su papel sobre la regulación de SHH y los factores de transcripción Msx1 y Msx2. Esta desregulación génica ocasiona diversas malformaciones como microftalmia, holoprosencefalia, labio y paladar hendido, así como hipoplasia de la línea media facial.16 Con certeza, podemos concluir que el estudio de éstos defectos al nacimiento mediante tecnología molecular, permitirán continuar profundizando en el conocimiento de los mecanismos génicos y celulares, que conllevan a un desarrollo facial normal, así como alteraciones en cualquiera de estas vías del desarrollo, conduce a malformaciones craneofaciales.
¿ Clasificación
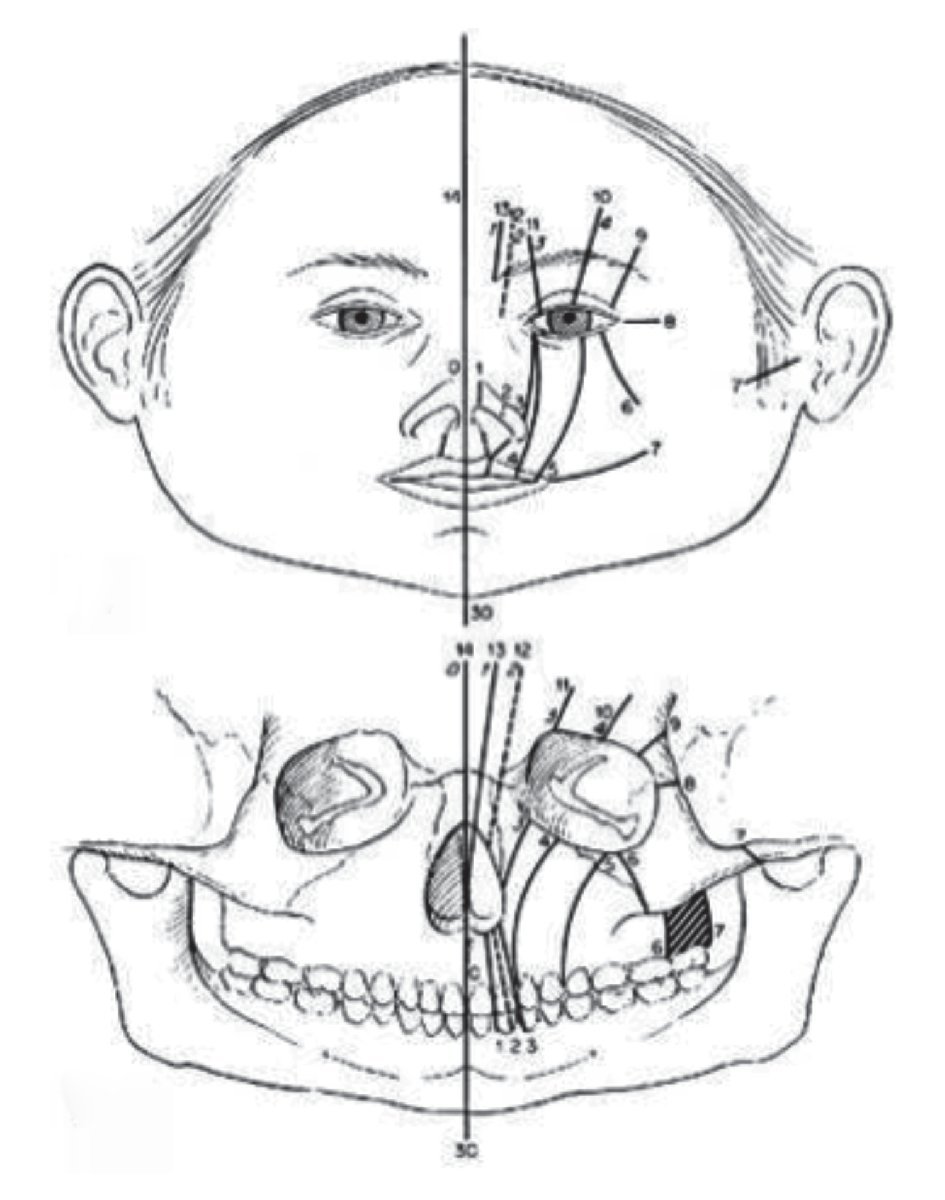
Las malformaciones craneofaciales pueden clasificarse de distinta maneras, de acuerdo a su etiología o a la región afectada. La clasificación de fisuras craneofaciales más reconocida en todo el mundo, debido a su amplia y extensa experiencia en el manejo y tratamiento en cirugía craneofacial, es la creada por el Doctor Paul Tessier. Esta clasificación, enumera las fisuras craneofaciales de 0 al 14, describe las características óseas y de tejidos blandos (Figura 1). El presente trabajo tiene como objetivo hacer una revisión del abordaje, diagnóstico y tratamiento de los principales síndromes asociados a malformaciones craneofaciales, ofreciendo una descripción clínica de los mismos.
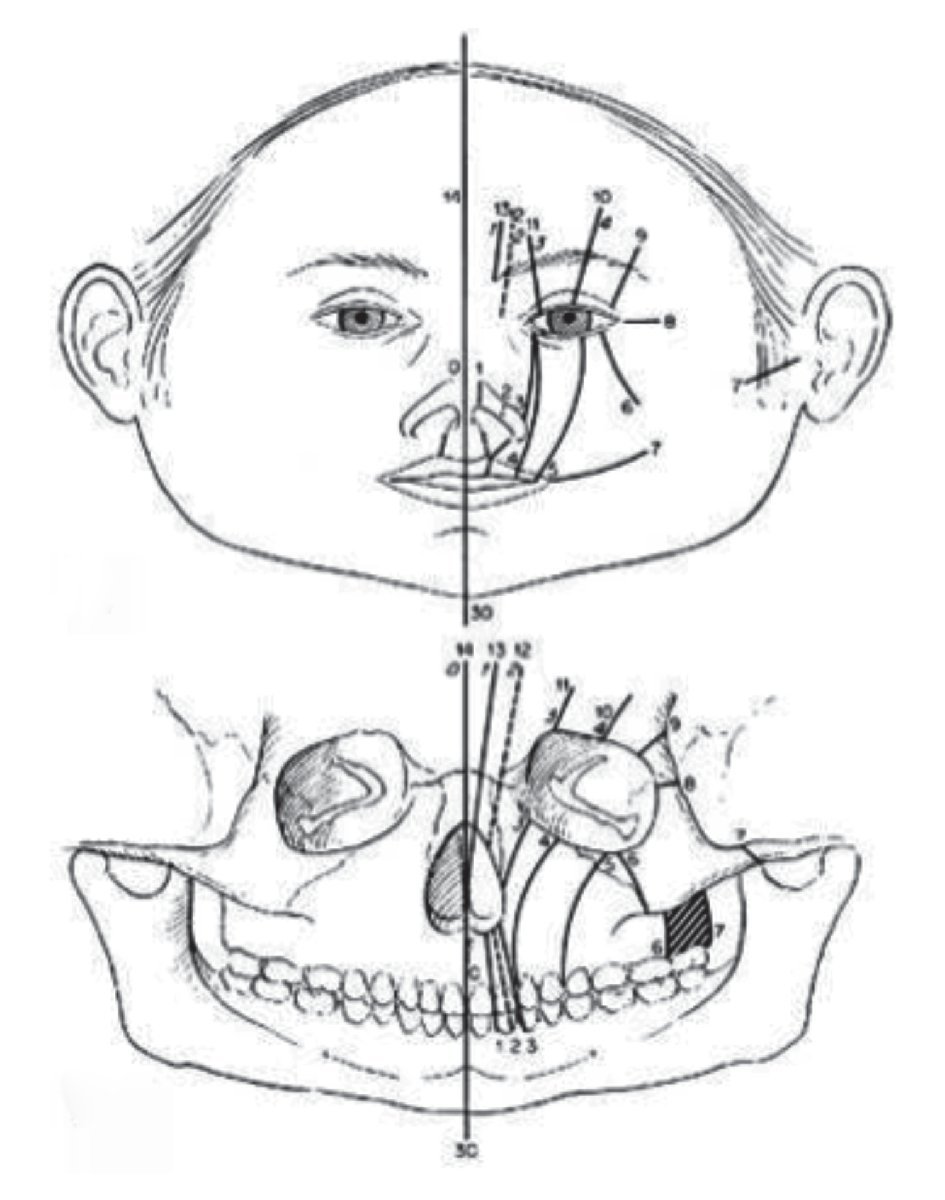
¿Figura 1. Clasificación de fisuras craneofaciales de Tessier. Hendiduras de línea media 0, 14. Hendiduras Paramedias 1, 2, 12 y 13. Hendiduras orbitarias 3, 4, 5, 9, 10 y 11. Hendiduras Laterales 6, 7 y 8. Tessier P. Anatomical classification facial, cranio-facial, and latero-facial clefts.
¿ Microsomía hemifacial
La microsomía hemifacial es un defecto al nacimiento, que involucra a los derivados del primer y segundo arco braquial. El espectro clínico fue más tarde extendido e incluye una serie de malformaciones oculares, auriculares y vertebrales, que fue renombrado como espectro óculo-aurículo-vertebral (OAVS), debido a la asociación con otras malformaciones cardiacas, vertebrales y del sistema nervioso central.
La microsomía hemifacial representa la forma más frecuente de asimetría facial aislada, ocurriendo en uno de cada 5 600 nacimientos.29 Es la segunda anomalía más común, después del labio y paladar hendido.30 Se trata de un síndrome del primer arco branquial, involucrando falta de desarrollo de la articulación temporomandibular, la rama mandibular, los músculos de la masticación y la oreja. El segundo arco branquial también puede estar involucrado, afectando entonces a su vez al nervio facial y a los músculos faciales.30 En el comienzo de la década de los sesenta, Gorlin describió un síndrome caracterizado primordialmente por microsomía hemifacial y fusión de vértebras, además de malformaciones óticas y oculares. A este síndrome se le llamó Síndrome de Goldenhar.31 En cuanto a la etiología de la microsomía hemifacial, autores opinan que puede tratarse de una pérdida temprana de células de la cresta neural,30 hemorragia fetal en los dos primeros arcos branquiales, ruptura y sangrado de la arteria estapedial.32,33 El lado derecho suele estar más afectado.34 La microsomía hemifacial parece ser más predominante en varones.33 Las características clínicas incluyen asimetría facial de severidad variable, microtia, línea media facial desviada hacia el lado afectado, arco zigomático corto e hipoplasia maxilar y temporal.36 A su vez, en pacientes con síndrome de Goldenhar se han descrito colobomas, hipoplasia de nervio óptico, hemivértebras y otras malformaciones de tubo digestivo, cardiacas, renales y respiratorias, por lo que el síndrome también es conocido como espectro OAVS.36 Se han identificado múltiples causas para la microsomía heía hea hemifacial, como factores ambientales, hereditarios, multifactoriales y desconocidos.
¿ Craneosinostosis
El esqueleto del cráneo humano consta de 22 huesos separados, comprende el viscerocráneo (mandíbula y otros derivados de los arcos braquiales), y el neurocráneo (base y bóveda craneana). Estos se forman durante la vida fetal, por medio de osificación endocondral y membranosa, los huesos planos dependen de osificación membranosa pero no se fusionan, dando lugar a una estructura funcional llamada sutura, que da la habilidad de adaptarse al crecimiento del cerebro. La región media de las suturas tiene células precursoras mesenquimales. En el humano se conocen seis suturas, dos coronales, una sagital, dos lamboideas y una metópica. Su fusión prematura ocasiona deformidades craneales, lo que se ha denominado craneosinostosis. El cierre temprano de estas suturas puede ocasionar aumento de la presión intracraneal, alteración del flujo sanguíneo, alteraciones de la visión y audición, así como alteraciones psicológicas o de retraso mental.
Las craneosinostosis se clasifican en sindromáticas (complejas) y no sindromáticas (simples).
La fusión temprana de las suturas puede ser secundaria a factores extrínsecos, tales como restricción intrauterina o administración de algunos fármacos durante el embarazo. Sin embargo, aproximadamente el 8% de las craneosinostosis sindromáticas y no sindromáticas, son familiares identificándose mutaciones en más del 20% de los casos. Actualmente, se conocen más de 100 síndromes con craneosinostosis. En ésta revisión mencionaremos aquellos síndromes asociados a mutaciones de los FGFRs (receptores de los factores de crecimiento de fibroblastos) y de TWIST.37
¿ Craneosinostosis producidas por mutaciones de FGFR
La familia de FGF (factores de crecimiento de fibroblastos), se expresa localmente durante la formación del hueso. En estadios tempranos, los genes FGFR1 y FGFR2 se expresan en células mesenquimales, durante la condensación del mesénquima previo a la deposición de la matriz ósea, sobre los huesos largos y las suturas craneales. En estadios tardíos y vida posnatal FGFR1 y FGFR2, se encuentran en preosteoblastos, así como en osteoblastos junto con FGFR3. En el momento, se conoce que la señalización producida por los FGF, regula la proliferación celular y su diferenciación.
Existen cuatro tipos de receptores para los FGF, todos ellos son receptores tirosin-cinasas y presentan una organización común, que es en heterodímeros con la capacidad de dimerizarse en presencia de su ligando, sufrir fosforilación y posteriormente, utilión y posteriormente, utilin y posteriormente, utilizar vías de señalización para que el mensaje llegue al núcleo. Muchas de las mutaciones que afectan a estos receptores, les confieren una activación constitutiva independiente de ligando (ganancia de función), que ocasiona osificación prematura de las suturas craneales.37
¿ Síndrome de Crouzon
Esta craneosinostosis tiene una frecuencia de uno en 50 000. Es causada por mutaciones del gen FGFR2 localizado en 10q26, y en pocos casos por mutaciones del gen FGFR3 en 4p16.37
Representa la craneosinostosis autosómica dominante más frecuente.18 Las manifestaciones incluyen hipodesarrollo de órbitas, con proptosis ocular consecuente.18 El estrabismo divergente de origen muscular también es frecuente.38 Existe maxilar hipoplásico y mala oclusión. Se ha reportado que la manifestación neurológica más común es la hipertensión intracraneal.18 Además, se asocia a complicaciones tales como queratitis, conjuntivitis, cefalea frecuente, hipoacusia conductiva y disminución de la agudeza visual.18,39 Se caracteriza por craneosinostosis coronal, sagital y lambdoidea.18
¿ Síndrome de Apert
El síndrome de Apert tienen una frecuencia de uno en 60000. Está ocasionado por mutaciones en FGFR2 localizado en 10q26.37 El síndrome de Apert también conocida con el nombre de acrocefalosindactilia tipo 1, es la craneosinostosis con manifestaciones más severas. Se caracteriza por hipoplasia de la línea media facial, proptosis, implantación baja de orejas, prognatismo mandibular y deformidad nasal.40 Existe sindactilia de segundo, tercer y cuarto dedos tanto en pies como manos.41 Las malformaciones de sistema nervioso central incluyen ausencia de septum pellucidum, hipoplasia de hipocampo, anormalidades de cuerpo calloso y displasia de corteza cereberal.42 Tales malformaciones se asocian a retraso mental. El cráneo posee un diámetro anteroposterior corto, originado por sinostosis coronal bilateral.18
¿ Síndrome de Pfeiffer
Tiene una frecuencia de uno en 100000. Se presenta por mutaciones en FGFR2 (locus 10q26) o FGFR1 (8p11).37 Se transmite con carácter autosómico dominante. Se han asociado mutaciones en el gen FGFR2.21 Se caracteriza principalmente por turricefalia.17 A nivel de extremidades, existen pulgares y primeros dedos de los pies cortos y desviados en sentido medial.18
¿ Craneosinostosis causadas por mutaciones del gen TWIST
Síndrome de Saethre-Chotzen
Con una frecuencia estimada de uno en 100 000, ocasionado por mutaciones en el gen TWIST localizado en 7p21, éste gen es un factor de transcripción con un dominio básico hélice-loop-hélice (HLH), que dimeriza y forma un complejo con la capacidad de unirse a secuencias consenso presentes en el promotor de sus genes blanco. Desempeña roles positivos y negativos, en la morfogénesis temprana. En estadios tempranos TWIST se requiere para la guía, migración y diferenciación de las células de la cresta neural, e indirectamente al expresarse en el mesénquima esquelético controla el desarrollo de las suturas, a través de la regulación de la proteína morfogenética del hueso (BMP) y la señalización del FGF, reprimiendo la proliferación de las células precursoras osteogénicas in vivo. Se ha observado que TWIST proporciona resistencia a la apoptosis, lo que representa un rol importante en el mantenimiento de las células. Las mutaciones en el dominio HLH del gen TWIST, ocasionan el aumento de la síntesis de colágeno tipo I, así como deposición de matriz extracelular resultando en incremento de osteogénesis, lo que explica la fusión prematura de las suturas. Se han encontrado mutaciones en estado heterocigoto, que ocasionan pérdida de la capacidad para unirse al DNA por lo que hay pérdida de función de la proteína o haploinsuficiencia. El síndrome de Seathre-Chotzen se caracteriza por alteraciones de los precursores osteogénicos, así como en la proliferación de éstas células y su sobrevida. Sin embargo, solo la sutura coronal se cierra precozmente en los pacientes con éste síndrome, lo que se ha tratado de explicar con una expresión diferencial en ésta sutura.37
Clínicamente se manifiesta por sinostosis coronal, aunque también puede haber sinostosis lambdoidea y metópica.18 El patrón de herencia también es autosómico dominante y entre otras manifestaciones faciales, se hallan la asimetría facial, hipertelorismo, hipoplasia maxilar, sindactilia, braquidactilia e implantación frontal del cabello baja.43 Este síndrome también es ampliamente conocido como acrocefalosindactilia tipo III.
Complejo malformativo de Pierre Robin
Un complejo malformativo se define como dos o más malformaciones vecinas, provenientes del mismo origen embrionario.44 El complejo se caracteriza por micrognatia mandibular o retrognatia, glosoptosis con o sin fisura del paladar. Ésta se ha reportado hasta en 90% de los casos.45-49 La incidencia se ha reportado de uno en 8500.48 En este caso, la hipoplasia mandibular representa la alteración primaria y se produce por deficiencia del mesénquima, en consecuencia existe mala posición de la lengua y ésta es la que origina, la falta de fusión de las conchas palatinas.44 Las principales manifestaciones clínicas son la obstrucción intermitente en la vía aérea superior y dificultad para la alimentación, suelen ser más frecuentes y severas en el periodo neonatal y los primeros meses de vida.44-48,50,51 Las causas de la secuencia de Pierre Robin no han sido esclarecidas, se ha sugerido la participación de teratógenos y además, puede formar parte de otros síndromes genéticos, como el síndrome de Sticker, síndrome velocardiofacial, displasias óseas entre muchos otros.
Síndrome de Treacher Collins
También conocido como disostosis mandíbulofacial, se trata de una entidad autosómica dominante, que afecta a las estructuras derivadas del primer y segundo arco branquial.52-55 No obstante, solo el 40% de los pacientes con síndrome de Treacher Collins tienen historia familiar positiva, el resto son debidos a mutaciones de novo. Se ha estimado una incidencia de uno en 50 000 nacidos vivos.56 Los criterios diagnósticos mayores del síndrome comprenden hipoplasia bilateral simétrica de la línea media facial, coloboma de párpado inferior, micrognatia, hipoplasia malar, microtia y otras malformaciones óticas, con el potencial de ocasionar hipoacusia conductiva.52
Es causado por mutaciones del gen TCOF1 localizado en 5q32-q33.1, el cual codifica para la proteína melasa localizada en el nucleólo. La proteína melasa está implicada en la biogénesis ribosomal, transcripción, nucleologenesis y tráfico de proteínas entre el núcleo y citoplasma, es requerida para la formación de células de la cresta neural y para la sobrevida de las células neroepiteliales. Las mutaciones de TCOF ocasionan haploinsuficiencia de la proteína melasa provocando pérdida de la habilidad de las proteínas, para trasportarse hacia el núcleo durante el desarrollo del primer y segundo arco braquial lo que provoca depleción de las células neurales troncales por apoptosis, afectando a su migración. Estudios claramente demuestran, que la hipoplasia craneofacial característica de éste síndrome es resultado directo de una deficiencia en el número de células de la cresta neural craneales.57,58
¿ Implicaciones clínicas de las malformaciones craneofaciales
La pérdida de función de la región afectada puede presentarse clínicamente con problemas para la deglución y la masticación, problemas de audición, visión y olfatorias, cefalea, anomalías sensitivas faciales y craneales, problemas de fonación entre otras. En este rubro nos enfocaremos a dos principalmente. Una de las complicaciones más importantes y que puede poner en riesgo la vida del paciente, es la obstrucción de la vía aérea superior como son la micrognatia, una posición anómala de la lengua, hipoplasia faríngea y laríngea y estrechamientos traqueales.53 Se han reportado diversos casos de pacientes que desarrollan hipoxia, hipercarbia, cor pulmonale e incluso muerte súbita.59 La atresia de coanas, puede causar asfixia en el neonato, provocar estridor y cianosis paroxística y por tal motivo, requieren de atención especializada en la reanimación neonatal.57 El manejo de la vía aérea en estos pacientes es un elemento crítico, dentro de la atención multidisciplinaria y debe ser individualizado. Estudios de nasofaringoscopía fibróptica a pacientes con malformaciones craneofaciales y apnea obstructiva, demuestran que existen cuatro tipos de obstrucción de vía aérea superior y el tipo 1, que resulta del desplazamiento posterior de la lengua provocando que ésta entre en contacto con la pared posterior de la faringe,51 es el tipo más común hallado en pacientes con el complejo de Pierre Robin,60-63 éste ocurre hasta en el 80% de los casos.62,63 En el caso de procedimientos anestésicos, se ha reportado el uso de máscara laríngea tanto en pacientes con Pierre Robin o Treacher Collins.64-66,68 Otros autores que han manejado pacientes con espectro OAVS, han optado por el uso de máscara laríngea flexible argumentando que ofrece la ventaja de evitar las manifestaciones asociadas a la extubación traqueal.69 En el caso del Síndrome de Apert, donde se han reportado anomalías en el cartílago traqueal,67,68 se ha reportado el uso exitoso de la broncoscopia rígida para el manejo de vía aérea.69 Uno de los objetivos del manejo es evitar la intubación endotraqueal prolongada, así como la realización de traqueostomía debido a las complicaciones inherentes de los procedimientos y los efectos a largo plazo. El manejo integral de los pacientes puede requerir del uso de la oximetría de pulso, videofluoroscopia de deglución y polisomnografía, ya que es factible el desarrollo de síndrome de apnea obstructiva del sueño.70
El otro punto que merece vigilancia estrecha es la dificultad para la alimentación y ésta se haya vinculada directamente a la respiración, ya que la dificultad para respirar origina incoordinación para succionar y para deglutir.51 Múltiples reportes demuestran que estos pacientes se encuentran en riesgo para el desarrollo de enfermedad por reflujo gastroesofágico, principalmente asociado al uso de tubos para alimentación.71-73 Un grupo multidisciplinario del Hospital para la Rehabilitación de Anomalías Craneofaciales de la Universidad de Sao Paulo, emplea una técnica para alimentación que permite promover la alimentación oral en un período de siete días y discontinuar el uso de la sonda nasogástrica.74 La técnica consiste en masaje para relajar y desplazar la lengua en sentido anterior, soporte manual para sostener la mandíbula, postura simétrica global y el uso de un biberón con un pezón con un agujero de 1 mm, el biberón se coloca con el pezón en modo preciso sobre la lengua y se efectúan movimientos rítmicos del pezón en la cavidad oral, mientras se alimenta al paciente, la sonda nasogástrica se retira una vez que el paciente acepta la alimentación oral.51 Otra estrategia empleada por este Hospital, es el uso de una dieta hipercalórica que consiste en leche materna o fórmula basada en leche, a la que se agregan de 5% a 8% de polímeros de glucosa, y de 3% a 5% de triglicéridos de cadena media.75,76
¿ Modalidades para el diagnóstico
Realizar un diagnóstico clínico es esencial. Identificar las malformaciones craneofaciales presentes, asociarlas entre sí, su repercusión clínica y realizar una exploración física completa a fin de descartar malformaciones sistémicas. El abordaje de éstos casos deberá estar conformado por un grupo multidisciplinario, que realice un estudio clínico exhaustivo, que incluya una evaluación genética, estudios de imagen y pruebas psicométricas. Dentro de los estudios de imagen, la radiografía simple puede ser útil para una evaluación inicial en donde se ubiquen alteraciones como sinostosis, en donde se observa ausencia de la lucidez normal en las suturas. Existen reportes de que la tasa de detección de malformaciones craneofaciales, en el período prenatal mediante ultrasonografía convencional es menor al 20%.77-79 Se ha estudiado y comparado, el empleo del ultrasonido 3D, el cual ha tenido mayor sensibilidad que el ultrasonido convencional.80-83 El uso del ultrasonido 3D supone entonces la ventaja de ofrecer una imagen más real de la cara del producto, y brindar mejor consejería a los padres para ayudarlos a comprender el padecimiento.84,85 La tomografía axial computada es una de las herramientas diagnósticas más ampliamente empleada y recomendada, ya que permite analizar el tejido óseo y blando. La ortopantomografía, la tomografía axial computarizada, tomodensitometría, TAC con reconstrucción 3D y resonancia magnética nuclear son estudios de gabinete con gran utilidad diagnóstica, ya que permiten definir con precisión las alteraciones y pueden ser de gran ayuda para el abordaje quirúrgico posterior.86-90 La cefalometría es otro adyuvante en el estudio de este tipo de pacientes.91 Permite estudiar el comportamiento de crecimiento craneofacial, permitiendo además elaborar estrategias de tratamiento o determinar pronóstico.92 En el 2007, se publicaron los resultados de un estudio retrospectivo cuyo objetivo era identificar las diferencias entre las proporciones verticales de los tejidos óseos y blandos, en pacientes con microsomía hemifacial sometidos a distracción ósea maxilomandibular simultánea.93 Determinaron las proporciones en radiografías y fotografías antes y después del tratamiento quirúrgico de 20 pacientes, 10 hombres y 10 mujeres con un rango de edad de los 12 a los 33 años. En cuanto al lado afectado, en 15 pacientes se presentó en el derecho y en cinco, en el izquierdo. Los cambios que se obtuvieron en el tejido blando, fueron mayores que en los óseos. Los autores concluyeron que la distracción ósea produce elongación tanto de tejidos blandos como óseos, aunque no en la misma proporción hallando que las asimetrías óseas y blandas no son idénticas.93
¿ Tratamiento
En 1950 Sir Harold Gillies, hoy considerado pilar de la cirugía plástica moderna, publicó un caso de oxicefalia, en el cual realizó un procedimiento quirúrgico diseñado para reponer mediante osteotomía, el componente del receso malar y maxilar del paciente. Los resultados fueron una mejor oclusión en los incisivos superiores, así como mejora de la proptosis de los ojos que presentaba el paciente.94 Actualmente, se considera que ésta fue la primera corrección quirúrgica exitosa de un caso de malformación craneofacial. Posteriormente, el cirujano francés Paul Tessier, conocido actualmente como el padre de la cirugía craneofacial, publicó los primeros trabajos donde describía osteotomías totales del macizo facial en pacientes con síndromes de Crouzon y Apert, así como en casos de oxicefalia, escafocefalia y turricefalia.92 Otras de sus aportaciones incluyen el describir osteotomías cráneo-naso-órbito-faciales, para hipertelorismo y el uso de la incisión coronal.95-97 México, es sin duda alguna uno de los países que más ha contribuido al desarrollo de la cirugía craneofacial en todo el mundo, con las grandes aportaciones del Doctor Fernando Ortíz Monasterio y la formación de la clínica de cirugía craneofacial. A su vez, otro gran especialista en la materia, el Doctor Antonio Fuente Del Campo, ha publicado una vasta cantidad de artículos sobre el tema con resultados exitosos. Ambos poseen un amplio número de publicaciones sobre los procedimientos quirúrgicos más empleados, actualmente en todo el mundo.98-106 Entre ellos, un trabajo pionero mundial publicado en 1978, un trabajo donde describían un procedimiento llamado "avance en bloque". Por otra parte, desde hace más de 10 años se ha empleado la distracción osteogénica mandibular, para el tratam iento de diversas malformaciones craneofaciales. La primera técnica fue descrita por Codivilla.107 Casi 70 años más tarde, Snyder experimentó empleando un distractor externo en la mandíbula de un modelo canino.105 Finalmente, fue a comienzos de década de los noventa que Karp realizó un estudio histológico seriado sobre la elongación de hueso membranoso, demostrando así la valiosa utilidad del procedimiento.108 En Estados Unidos, McCarthy publicaría un estudio similar con resultados favorables.109,110 En términos generales, debido a que pretendemos ofrecer únicamente una breve reseña sobre la cirugía, la distracción es una técnica que permite la regeneración de hueso nuevo entre dos segmentos óseos vascularizados y que son gradualmente separados por fuerzas mecánicas.38 De ésta manera, puede resolverse el dilema que supone tratar a pacientes con diversos síndromes, por ejemplo Crouzon, Pierre Robin o microsomía hemifacial. Grandes expertos internacionales como Molina en México, McCarthy en Estados Unidos y Diner en Francia, han obtenido resultados extraordinarios. Precisamente, hace más de una década que en México, se han publicado resultados satisfactorios y con seguimiento a largo plazo, en el área de la cirugía craneofacial.111 Se ha acumulado una vasta experiencia en la distracción ósea, para el tratamiento de malformaciones craneofaciales en sus modalidades externa, interna y modificada por diversos cirujanos.111-114 Los tres períodos secuenciales comúnmente reconocidos de la distracción osteogénica son la latencia, distracción y consolidación. La primera se extiende desde la primera osteotomía hasta la tracción del hueso, con lo que se inicia la formación de callo óseo. Posteriormente, en la distracción se advierta la evolución del tejido de granulación a la sustitución por tejido fibroso y finalmente la formación de hueso, para éste momento se ha evolucionado del callo blando al callo duro. Finalmente, en la fase de consolidación la función expansiva del distractor ha cesado y se han formado corticales óseas, que permiten la permanencia de la elongación del hueso. Se ha publicado también sobre la distracción ósea asistida endoscópicamente, como una técnica novedosa y prometedora.38 La distracción ósea posee la ventaja de ser menos invasiva, que otros accesos en cráneo y macizo facial y supone una menor pérdida sanguínea y menor tiempo quirúrgico. Del mismo se ha demostrado buena fusión ósea, con el crecimiento del nuevo hueso inducido.115 Existen múltiples reportes, sobre el uso de la distracción ósea en este tipo de pacientes alrededor del mundo y la experiencia y aceptación continúa en ascenso.116-139 En todo caso, el tratamiento de los pacientes debe ser individualizado y sometido a una exhaustiva planeación, sobre el procedimiento a realizar.
¿ Conclusiones
En base a lo anterior, podemos finalizar haciendo énfasis en la importancia de un abordaje multidisciplinario que garantice un diagnóstico certero. Además de un manejo, que permita un resultado funcional y estético satisfactorio para el paciente, siempre con apoyo psicológico para el manejo de posibles secuelas. El equipo de trabajo deberá estar constituido por especialistas en genética médica, cirugía plástica, reconstructiva y craneofacial, pediatría, neurocirugía, neurología, otorrinolaringología, foniatría, oftalmología, neonatología, cirugía maxilofacial, odontología, ortodoncia, psiquiatría, nutriología, patología del lenguaje, rehabilitación y psicología, entre otros. El conocimiento de los mecanismos genéticos que conducen al desarrollo craneofacial normal, permitirá identificar aquellos genes que al alterarse ocasionan diversos padecimiento, de tal manera que puedan desarrollarse a su vez nuevas herramientas de diagnóstico molecular. Cabe señalar, que el servicio de Genética del Hospital General de México ha contribuido en colaboración, con diversos servicios en el manejo de éste grupo de pacientes.
Correspondencia: Dr. Damián Palafox.
San Francisco No. 7. Col. Del Valle. C.P. 03100. Delegación Benito Juárez. México D.F., México.
Correo electrónico:palafoxdamian@yahoo.fr