



El síndrome poliglandular autoinmune (SPA) es una rara entidad caracterizada por la asociación de por lo menos dos insuficiencias glandulares mediadas por mecanismos autoinmunes. La clasificación inicial de este síndrome fue ampliada para describir cuatro tipos bien definidos de hipofunción glandular, desde aquellas que se presentan en la infancia (SPA tipo 1), como las que usualmente tienen su presentación en la edad adulta (SPA tipo 2, SPA tipo 3). En la presente revisión se señalan aspectos epidemiológicos, de clasificación, genética, clínica e identificación de anticuerpos dirigidos contra glándulas diana, así como el tratamiento de dicha entidad.
Autoimmune polyglandular syndrome (APS) is an uncommon entity characterized by the association between two glandular insufficiencies mediated by autoimmune mechanisms. The initial classification was extended to describe four types of glandular hipofunction, from those occurring in childhood (APS type 1), to those that occurred during the adult age (APS type 2, APS type 3). This review presents epidemiologic, classification, genetic and clinic aspects, as well as the classifications of autoantibodies directed against target glands, and the treatment of this disease.
• Introducción
Bajo la determinación del síndrome poliglandular autoinmune (SPA) se define el raro desorden caracterizado por la coexistencia de por lo menos dos insuficiencias glandulares mediado por mecanismos inmunitarios, conjuntamente con ello otras enfermedades autoinmunes no endocrinológicas pueden coexistir.1,2
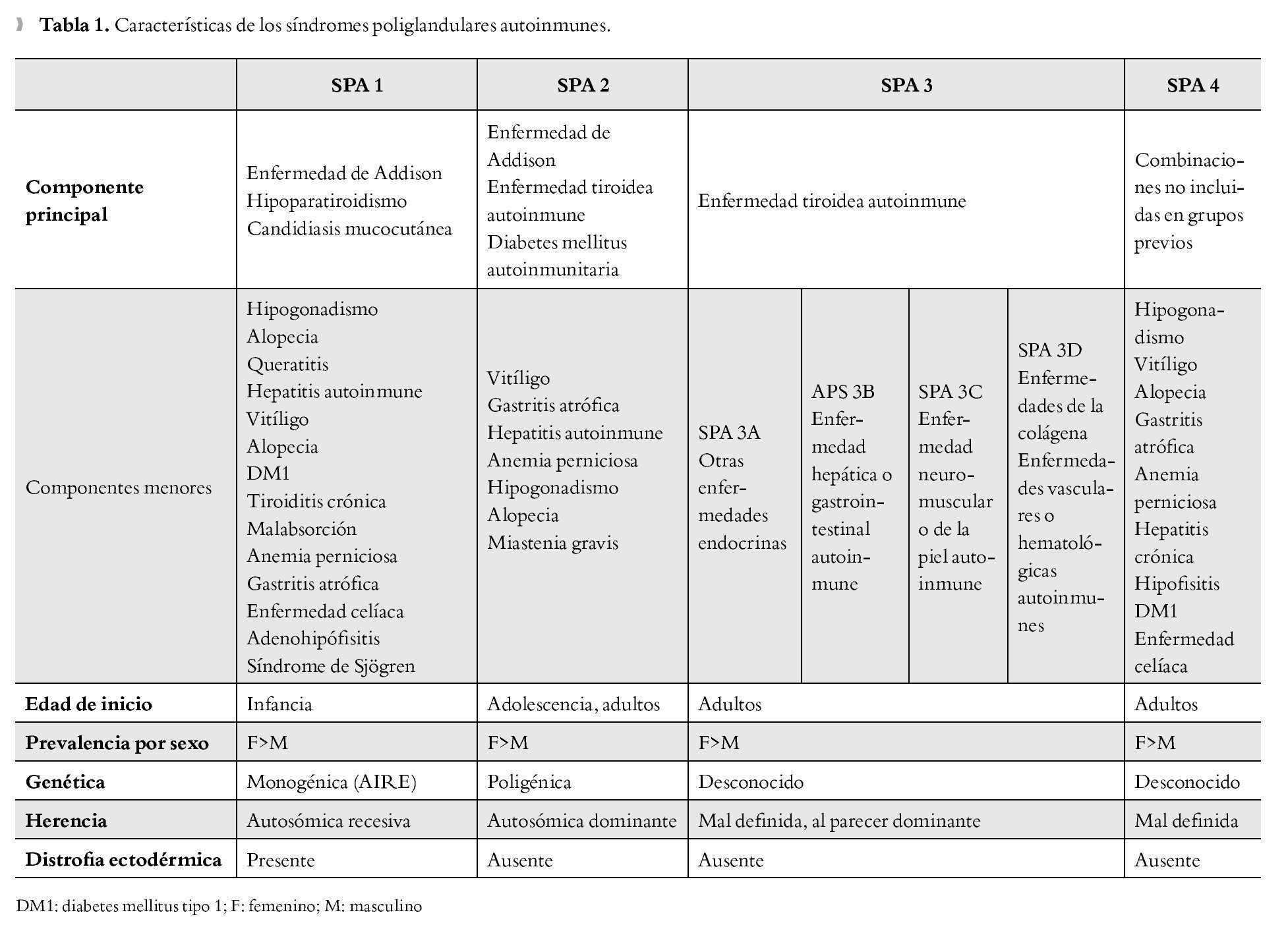
Es importante para el médico el reconocimiento clínico del SPA (Tabla 1) ya que la falta de éste puede llevar al subdiagnóstico, en este sentido la detección precoz llevaría a un adecuado remplazo hormonal, el cual es de vital importancia particularmente cuando se asocia a insuficiencia adrenal o tiroidea. Una identificación rápida también llevaría a seguimiento familiar estrecho, dado el patrón de herencia que presentan variantes de dicho síndrome. Por último, es importante estar alerta ante la posibilidad de un fallo endocrino secundario, por lo se justifica la búsqueda de disfunciones endocrinológicas y no endocrinológicas posteriores.
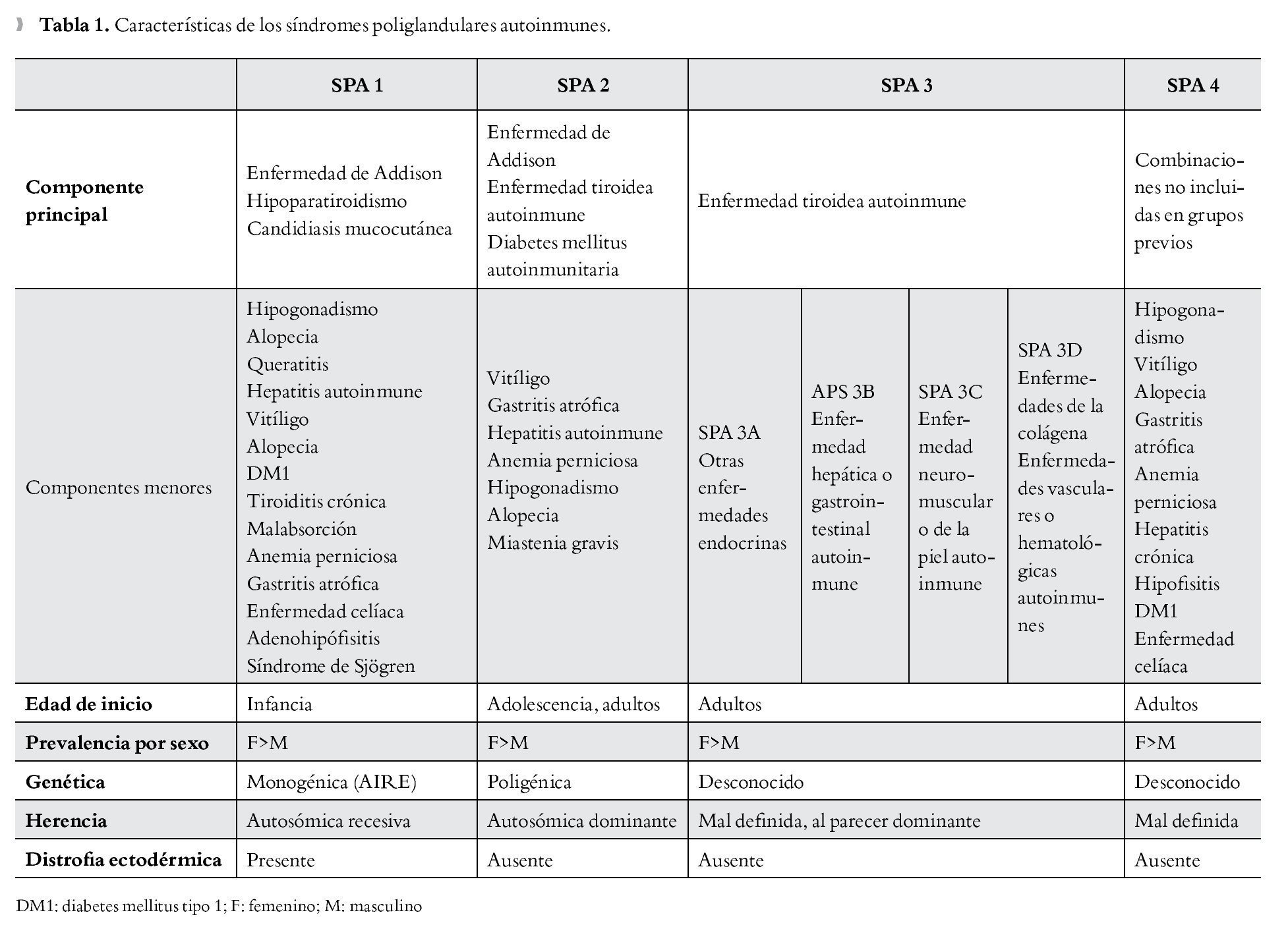
Fue Addison médico británico quien en 1855,3 describió la enfermedad que posteriormente llevaría su nombre, caracterizada por deficiencia de hormonas esteroideas. Thorpe y Handley en 1929 describieron la asociación entre hipoparatiroidismo y candidiasis mucocutánea;4 Leonard5 en 1946 asocia ya el hipoparatiroidismo con insuficiencia suprarrenal (ISR). En 1926, Schmidt describió en dos pacientes la asociación de tiroiditis linfocítica crónica e ISR de origen no tuberculoso; Carpenter en 1964 amplió la descripción con la asociación de diabetes mellitus autoinmunitaria; posteriormente en 1981 Neufeld y Blizzard realizan la clasificación inicial de los SPA,1 definiendo dos grandes grupos: el SPA tipo 1, que asociaría la ISR con el hipoparatiroidismo y el SPA tipo 2, más frecuente, estaría constituido por la ISR con la diabetes mellitus tipo 1 (DM1) y/o la enfermedad tiroidea autoinmune, modificaciones posteriores de dicha clasificación inicial llevarían a describir cuatro tipos de SPA (Tabla 2); el tipo 3 que generalmente se presenta en la edad adulta y el cual se diferencia del tipo 1 y 2 por la falta de afección a nivel suprarrenal, y el tipo 4 en donde están presentes dos o más enfermedades autoinmunes órgano-específicas que no cumplan criterios del tipo 1, 2 o 3. Eisenbarth y Gottlieb6 en 2004, abrieron el debate en cuanto a la clasificación de los SPA, recomendando la denominación de sólo dos tipos, 1 y 2, justificando que la división en cuatro subtipos llevaría a un nombre inapropiado, ya que estos síndromes podrían incluir una serie de trastornos no endocrinológicos.

• Inmunidad
La enfermedad glandular autoinmune afecta a individuos genéticamente susceptibles, en los cuales hay demostración de infiltración linfo-monocitaria. Esta afección se produce como consecuencia de la pérdida de inmunotolerancia frente a autoantígenos, pudiendo ser los mecanismos de esta pérdida la alteración en supresión de células autorreactivas o la presencia de antígenos extraños al organismo.7 Se ha sugerido que el desarrollo de autoinmunidad múltiple en el contexto del SPA, puede ser debido a uno o varios epítopes compartidos como agentes ambientales o como un autoantígeno común presente en varios tejidos endocrinos.8 Una hipótesis señala que órganos derivados de la misma capa germinal expresarían autoantígenos específicos, que tendrían como objetivo dichos órganos diana. Según esta teoría, el SPA tipo 2 sería resultado de afectación de capas distintas, el mesodermo (corteza adrenal) y endodermo (glándula pancreática y/o tiroidea), por dos autoantígenos diferentes o por un solo autoantígeno común a ambas capas.9 Con base a lo anterior, los SPA se caracterizan por una etapa potencial en donde se detectan anticuerpos circulantes contra células diana pero sin alteración funcional, posteriormente una etapa subclínica de infiltración mononuclear preferentemente linfocítica con destrucción glandular progresiva y en donde se puede encontrar alteraciones en pruebas funcionales, sucedida por una etapa final con manifestaciones clínicas bien definidas que corrobora la insuficiencia glandular.
El diagnóstico de SPA sólo se establece una vez corroborada la insuficiencia glandular, sin embargo la detección de anticuerpos en fases iniciales antes de la presentación clínica de la enfermedad puede predecir un SPA latente, en este sentido se han descrito posibles factores externos como desencadenantes en pacientes genéticamente susceptibles, originando la aparición brusca de una deficiencia glandular.8
La destrucción autoinmunitaria progresiva de la función glandular en el estadio final de la enfermedad, conlleva sustitución de tejido normal por tejido fibroso quedando así por ejemplo, la glándula suprarrenal con un peso aproximado de solo 1 g, comparado con su peso normal 3 a 5 g y diferenciándose de la ISR de causa tuberculosa, porque en esta última se suelen encontrar masas y calcificaciones.10
• SPA tipo 1
También conocido como síndrome de poliendocrinopatía autoinmune-candidiasis-distrofia ectodérmica (APECED, por sus siglas en inglés), es definida como la asociación de por lo menos dos de los tres componentes principales a saber: candidiasis mucocutánea crónica, hipoparatiroidismo primario e ISR.1,11 Perheentupa reporta la tríada completa sólo en un 50% de sus pacientes,12 y se sugiere que entre más temprano aparezca el primer componente del síndrome, mayor es la posibilidad de que aparezca un número mayor de los mismos.11,13 De forma característica, el síndrome aparece en infantes teniendo generalmente una aparición en secuencia cronológica, iniciando con candidiasis crónica generalmente antes de los cinco años, pudiendo aparecer incluso antes del mes de edad, el hipoparatiroidismo antes de los 10 años e ISR alrededor de los 15 años.11,13,14
Aunque es un síndrome raro se han reportado mayor prevalencia entre poblaciones con alto grado de consanguineidad. La prevalencia varía entre distintas regiones, siendo las más afectas los judíos iraníes (1:6 500 a 9 000),15 en la región de Cerdeña Italia (1: 14 400)16 y Finlandia (1:25 000).11 La relación entre mujer/hombre también varía de 0.8/1 a 2.4/1 según distintas series.11,13
El SPA tipo 1 se asocia a un patrón de herencia autosómico recesivo, debido a variadas mutaciones del gen regulador autoinmune AIRE, el cual es un factor de transcripción humano localizado en el cromosoma 21q22.3, compuesto de 14 exones y con un peso molecular aproximado de 58 kDa.17 El gen AIRE es expresado principalmente en las células epiteliales tímicas medulares, aunque también se expresa en menor medida en ganglios linfáticos, bazo, médula ósea, glándulas suprarrenales, hígado, células de sangre periférica. Se postula que dicho gen juega un papel preponderante en la inducción a la tolerancia de células T, a través de la expresión de autoantígenos periféricos en las células epiteliales del timo, en la selección negativa hacia dichas células autorreactivas, en la regulación de la transcripción y en el proceso tímico de inducción y mantenimiento de la autotolerancia inmunológica.17
Aunque preponderantemente el síndrome se asocia a patrón de herencia autosómico recesivo, se ha reportado un caso de herencia autosómico dominante asociado a la mutación G228W.18 Dentro de las más de 60 mutaciones del gen AIRE descritas hasta ahora, sólo pocas son de relevancia dada su frecuencia como la mutación R257X en el exón 6, encontrada hasta en un 80% de los casos en pacientes finlandeses, la mutación Y85C en el exón 2 frecuente entre judíos iraníes, la mutación R139X presente en el exón 3 común en Cerdeña y la mutación del 13 en el exón 8 encontrada en pacientes europeos del norte-occidente, británicos, así como sus descendientes americanos.16,19-21
Asimismo, se ha reportado cierta correspondencia con rasgos fenotípicos entre distintas mutaciones; la R257X asociada a candidiasis mucocutánea y baja prevalencia de ISR, así como la queratopatía entre judíos iraníes con la mutación Y85C, entre otras.15
• Clínica
El síndrome se presenta con una asociación de deficiencias glandulares de origen autoinmunitario que se preceden unas de otras tras meses o años, en algunas ocasiones se asocian a entidades inmunitarias no endocrinológicas.
La infección superficial crónica por Cándida albicans está presente en casi la totalidad de los pacientes con SPA tipo1,22 siendo menos prevalente entre judíos iraníes,15 pudiéndose presentarse desde el primer mes de vida hasta los 21 años, con media de edad de ocurrencia a los 6.5 años.13 Comúnmente afecta menos del 5% de la superficie corporal,11 predominantemente mucosa oral provocando grados variables de inflamación de la misma, incluso manifestándose como candidiasis hiperplásica crónica con placas blanquecinas hiperqueratósicas, o bien la forma atrófica con mucosa delgada y leucoplasia, la cual posee potencial carcinogénico.23 Otros lugares comunes de presentación de la infección candidiásica son a nivel del esófago provocando dolor retroesternal, piel y anexos, en donde la infección de las uñas generalmente precede a la de la boca; también se ha visto a nivel intestinal provocando meteorismo y/o diarrea y como vulvovaginitis.23 Se considera que la aparición de candidiasis mucocutánea en el SPA tipo1, es debido a un déficit inmunológico de hipersensibilidad retardada contra antígenos de Cándida albicans, catalogándose como una inmunodeficiencia celular secundaria,24 manteniendo intacta la respuesta humoral, lo que prevendría de la candidiasis sistémica.
El hipopatiroidismo generalmente es la primera anormalidad endocrina que aparece en el SPA tipo 1, está reportado en aproximadamente el 79% a 100% de los pacientes,10,11,25 pudiéndose presentar desde los 19 meses hasta los 44 años,11 con promedio de aparición a los 7.5 años,13 afecta preferentemente al sexo femenino26 y raramente cuando se presenta en la edad neonatal es crucial distinguirlo de enfermedades genéticas como el síndrome de DiGeorge, enfermedad de Kenney-Caffey o el síndrome de Barakat, que cursan con hipocalcemia persistente.10
Su traducción clínica depende del grado de hipocalcemia existente, desde la fase asintomática hasta contracturas musculares paroxísticas y/o eventos convulsivos secundarios a hipocalcemia. En el examen físico se hacen evidentes los signos de Chvostek (espasmo facial, especialmente de la comisura labial al percutir el nervio facial por delante de la oreja) y Trousseau, más específico, que consiste en la aparición de espasmo doloroso carpo pedal al reducir la circulación del antebrazo mediante la compresión con el brazalete del tensiómetro, por encima de la presión sistólica durante al menos tres minutos.
La ISR en el SPA tipo 1 aparece en el 60% a 100% de los casos,10,13 con presentación promedio a los 14.6 años,27 se reconoce clínicamente hasta cumplirse destrucción glandular superior al 90% y se manifiesta por una constelación de síntomas poco específicos como fatiga, debilidad generalizada que mejora con el reposo, mareo, síntomas gastrointestinales como náusea, calambres abdominales, vómito y diarrea, pérdida de peso; también puede presentarse hiperpigmentación de piel y mucosas, sobre todo en áreas previamente pigmentadas como mamas y genitales externos, áreas expuestas al sol o con microtraumas, dicha pigmentación se presenta como resultado de elevación plasmática de propiomelanocortina, precursor de la lipotropina b, corticotropina y melatonina, es reconocido como el signo más específico de la ISR primaria.10 También suele observarse sintomatología por deficiencia mineralocorticoide como hipotensión preponderadamente ortostática, deseo por la sal, trastornos hidroelectrolíticos como hiponatremia, hiperkalemia e hipercalcemia. Otras anomalías encontradas son decremento de la líbido, ligera acidosis, hipoglucemia, eosinofilia leve, linfocitosis y anemia normocítica ligera.28 En dado caso de no tratarse la ISR puede empeorar y ante un factor estresante como infección o evento quirúrgico, desencadenar una crisis suprarrenal.
La demostración de la insuficiencia se realiza midiendo las concentraciones séricas del cortisol y hormona adrenocorticotropa (ACHT) usualmente entre las 6:00 y 8:00 horas, una concentración de cortisol menor de 3 μg/dL (83 nmol/L) es indicativa de esta enfermedad, mientras que concentraciones mayores de 18 μg/dL la excluyen.28 La determinación de ACTH basal también ayuda a diferenciar entre ISR primaria de la secundaria, dado que en la primera los valores de ACTH suelen encontrase usualmente por encima de 100 pg/mL, mientras en la forma secundaria se encuentran bajos o inapropiadamente normales. En pacientes con sospecha de la enfermedad y prueba inicial no confirmatoria, se realiza una serie de pruebas dinámicas de estimulación glandular.28 En estudios de imagen de resonancia magnética (RM) o tomografía axial computarizada (TAC), muestran glándulas suprarrenales normales o atróficas.27 Es importante señalar que cuando se presenta la ISR como primera endocrinopatía en el contexto de SPA tipo 1, la susceptibilidad para presentar hipoparatiroidismo se reduce.26
• Componente menores del SPA tipo 1
Hipogonadismo hipergonadotrófico afecta mayoritariamente al sexo femenino pudiendo aparecer en un 24% a 60% en el SPA tipo 1,10 se reconoce clínicamente como amenorrea primaria, trastornos del desarrollo puberal o falla ovárica prematura. Al igual que las demás glándulas afectadas por el síndrome, histológicamente la gónada se encuentra con hipoplasia, fibrosis e infiltración linfocitaria.29
El desarrollo de enfermedad tiroidea autoinmune dentro del SPA tipo 1 es poco común, con una edad promedio de presentación de 20.3 años,27 siendo más frecuente la asociación de hipotiroidismo que hipertiroidismo, este último casi anecdótico. Se ha observado una mayor frecuencia de enfermedad tiroidea, con la mutación específica G228W del gen regulador autoinmune en el exón 6 en estado heterocigoto.18
Otros desordenes endocrinos que se pueden presentar, sin bien con mucha menor incidencia dentro del SPA tipo 1, son la DM1 en el 0% a 12%,27 los casos raros de hipofisitis linfocítica que afectan predominantemente la hormona de crecimiento (GH), seguida por vasopresina y deficiencia de ACTH.30
Dentro de los trastornos no endocrinológicos se encuentran la gastritis atrófica crónica (13% a 27%),27 con desarrollo ulterior de anemia perniciosa (0% a 15%),13,27 hepatitis autoinmune, el cual es descrito en un 10% a 15% de los pacientes con SPA tipo 1,31 y edad de inicio de cinco a 21 años27 presentándose ésta, con grados variables de severidad desde hepatitis leve hasta cuadros fulminantes. También pueden encontrarse cuadros de bronquiolitis o bronquiectasias autoinmunitarias, distrofia ectodérmica caracterizado por la tríada de queratoconjuntivitis, hipoplasia del esmalte dental y defectos ungueales.11 El vitíligo está presente en el 8% a 25%, la alopecia en el 13% a 72%,10 vasculitis, asplenia,32 atrofia del nervio óptico,33 colelitiasis, anemia hemolítica, colelitiasis, neoplasias malignas como el carcinoma epidermoide de la mucosa oral, lengua, esófago y adenocarcinoma gástrico,10,13,27 también han sido reportadas.
• SPA tipo 2
Se constituye por la coexistencia de dos o más deficiencias glandulares de origen autoinmune, en las que se encuentran afectadas la glándula suprarrenal y la glándula tiroidea (síndrome de Schmidt) o la glándula suprarrenal y el páncreas (síndrome de Carpenter), esto como componentes mayores pudiendo también presentarse insuficiencia gonadal, enfermedad celíaca, miastenia gravis y otros padecimientos autoinmunes no endocrinológicos, como en el SPA tipo 1 antes descrito. Betterle10 encontró presencia de tríada clásica de enfermedad de Addison, enfermedad tiroidea y DM1 en tan sólo 11% de 107 pacientes estudiados y al igual que en los demás tipos de SPA, la enfermedad está precedida por una etapa preclínica con elevación de anticuerpos órgano específicos. Tiene una incidencia de 1.4 a 4.5 casos en 100 000 habitantes,27 afectando preferentemente al sexo femenino sobre todo en la asociación de enfermedad de Addison y enfermedad tiroidea. Su presentación suele ocurrir hacia la tercera o cuarta década de la vida,1,34 siendo muy raro en la infancia.
La presencia de ISR se encuentra en el 89% a 100% de los pacientes, seguida de 69% a 82% de enfermedad tiroidea y 30% a 52% de DM1.10,27 Contrastando con lo anterior, Dittamar35 en un seguimiento por 13 años de 151 pacientes con SPA tipo 2, encontró a la DM1 como el componente endocrino más frecuente del síndrome (60.9%), mientras que la ISR y falla gonadal fueron pocos frecuentes con 18.5% y 5.3%, respectivamente. Dentro de la enfermedad tiroidea en el SPA tipo 2, la enfermedad de Graves se desarrolla antes de la ISR, la tiroiditis linfocítica crónica suele presentarse coincidentemente o posterior a la falla adrenal, mientras que el diagnóstico de diabetes mellitus autoinmune suele preceder a la enfermedad de Addison, esto generalmente en pacientes jóvenes.36,37 El lapso de presentación entre la primera y segunda endocrinopatía varía considerablemente, con un largo intervalo entre diabetes tipo 1 y enfermedad tiroidea (13.3 +/- 11.8 años), y corto periodo entre enfermedad de Adisson y enfermedad tiroidea autoinmune,35 parece que cuando la enfermedad tiroidea se presenta como primer componente del síndrome, el intervalo hasta la presentación de la segunda inmunopatía suele ser relativamente corto.35
Al igual que en el SPA tipo 1, otras enfermedades autoinmunitarias pueden verse en menor medida como el hipogonadismo hipergonadotrófico (4% a 9%), vitíligo (4.5% a 11%), alopecia (1% a 4%), hepatitis inmunitaria (4%), gastritis atrófica y anemia perniciosa (4.5% a 11%).27 También mucho más raro, se ha descrito la asociación del SPA tipo 2 con hipertensión arterial pulmonar primaria.38
Se piensa que en el SPA tipo 2 existe una predisposición de múltiples genes del cromosoma 6, el cual juega un rol predominante a través de su relación con el antígeno leucocitario humano (HLA) y que en asociación con factores ambientales desencadenaría el proceso autoinmune. Estudios iniciales relacionaron la aparición de la enfermedad con alelos B8 y DR3 del HLA.39 También se ha relacionado la aparición de ISR dentro del SPA tipo 2 con alelos dentro del HLA-DR3 con haplotipos DRB1*0301, DQA1*0501 y DQB1*0201.40,41 Consecuencia a su patrón de herencia autosómico dominante con penetrancia incompleta, los familiares de estos pacientes pueden ser afectos al síndrome con anterioridad, por lo tanto se recomienda realizar periódicamente exámenes de detección entre familiares de pacientes afectados.
• SPA tipo 2 incompleto
Betterle10 propuso el término de SPA tipo 2 incompleto, para clasificar a los pacientes que teniendo ISR presentaban además anticuerpos anti-tiroideos y/o anticuerpos para diabetes autoinmunitaria positivos como ICA o anti-GAD65 o bien, si presentaban enfermedad tiroidea autoinmune y/o DM1 mostraban también anticuerpos positivos antiadrenales (AA, 21-OH), sin evidencia de insuficiencia glandular e independiente del antecedente familiar. Asimismo, dependiendo de la situación funcional de la glándula, el SPA tipo 2 incompleto puede dividirse a sus vez en forma potencial o subclínica.10,27 Pacientes con antecedente de ISR y anticuerpos antitiroideos y/o para diabetes autoinmunitaria positivos pero sin alteraciones en las pruebas de estimulación glandular como curva de tolerancia a la glucosa deben ser catalogados como potenciales para SPA tipo 2; en el mismo rubro se clasificarían los pacientes con DM1 y/o con enfermedad tiroidea autoinmunitaria que presenten ACA/21-OH positivos con función adrenal normal. En cambio son considerados con SPA tipo 2 incompleto forma subclínica, a aquellos pacientes que presenten evidencia de una insuficiencia glandular aunado a anticuerpos positivos contra otra célula diana y además, presenten pruebas de estimulación anormales a dicha glándula.27 Entre las diferentes combinaciones que caerían dentro de esta categoría estarían pacientes con enfermedad de Addison y anticuerpos antitiroideos, que presenten además hipotiroidismo o hipertiroidismo subclínico o pacientes con enfermedad de Addison con ICA o anti-GAD65 presentes y, además intolerancia a la glucosa posterior a prueba de sobrecarga oral de glucosa. El seguimiento de los pacientes con SPA tipo 2 incompleto tanto potencial como subclínico a través de pruebas funcionales de estimulación, sería fuertemente recomendado para prevenir el brote de la enfermedad autoinmune o para otorgar un tratamiento oportuno de la deficiencia.
• SPA tipo 3
Se define como la asociación entre la enfermedad tiroidea autoinmune con otras entidades mediadas por mecanismos autoinmunes, y que según la reclasificación de Betterle27 puede subdividirse a su vez en:
SPA tipo 3A, enfermedad tiroidea asociada con enfermedades endocrinológicas como la DM1, síndrome de Mirata, hipofisitis linfocitaria, neurohipofisitis o falla ovárica prematura.
SPA tipo 3B, enfermedad tiroidea asociada a enfermedades gastrointestinales o hepáticas como anemia perniciosa o gastritis atrófica, enfermedad celíaca, enfermedad inflamatoria intestinal crónica, hepatitis autoinmune, cirrosis biliar primaria o colangitis esclerosante.
SPA tipo 3C, enfermedad tiroidea asociada a enfermedad de la piel o neuromuscular como el vitíligo, alopecia, miastenia gravis, esclerosis múltiple, síndrome del hombre rígido, trombocitopenia autoinmune, anemia hemolítica autoinmune y síndrome antifosfolípido.
SPA tipo 3D, enfermedad tiroidea asociada a enfermedades de la colágena y vasculitis como lupus eritematoso sistémico o discoide, artritis reumatoide, artritis reactiva, esclerodermia, síndrome de Sjögren, vasculitis.
• SPA tipo 4
Este tipo mucho más raro que los anteriores es la asociación de dos o más enfermedades autoinmunes órgano-específicas no incluidas dentro de la categoría de los tipos 1, 2 y 3. Esto puede incluir la asociación de ISR con los componentes menores de enfermedades autoinmunitarias como hipogonadismo, vitíligo, alopecia, hepatitis inmunitaria, gastritis atrófica y anemia perniciosa, etc. La determinación de anticuerpos positivos contra glándulas tiroidea y pancreática y/o posterior aparición de deficiencia glandular, permitiría reclasificarlos nuevamente como variantes del SPA tipo 2. En forma similar a los tipos 1 y 2 del SPA, la atrofia de la glándula suprarrenal se haría evidente por medio de estudios de imagen generalmente, RM.
• Anticuerpos
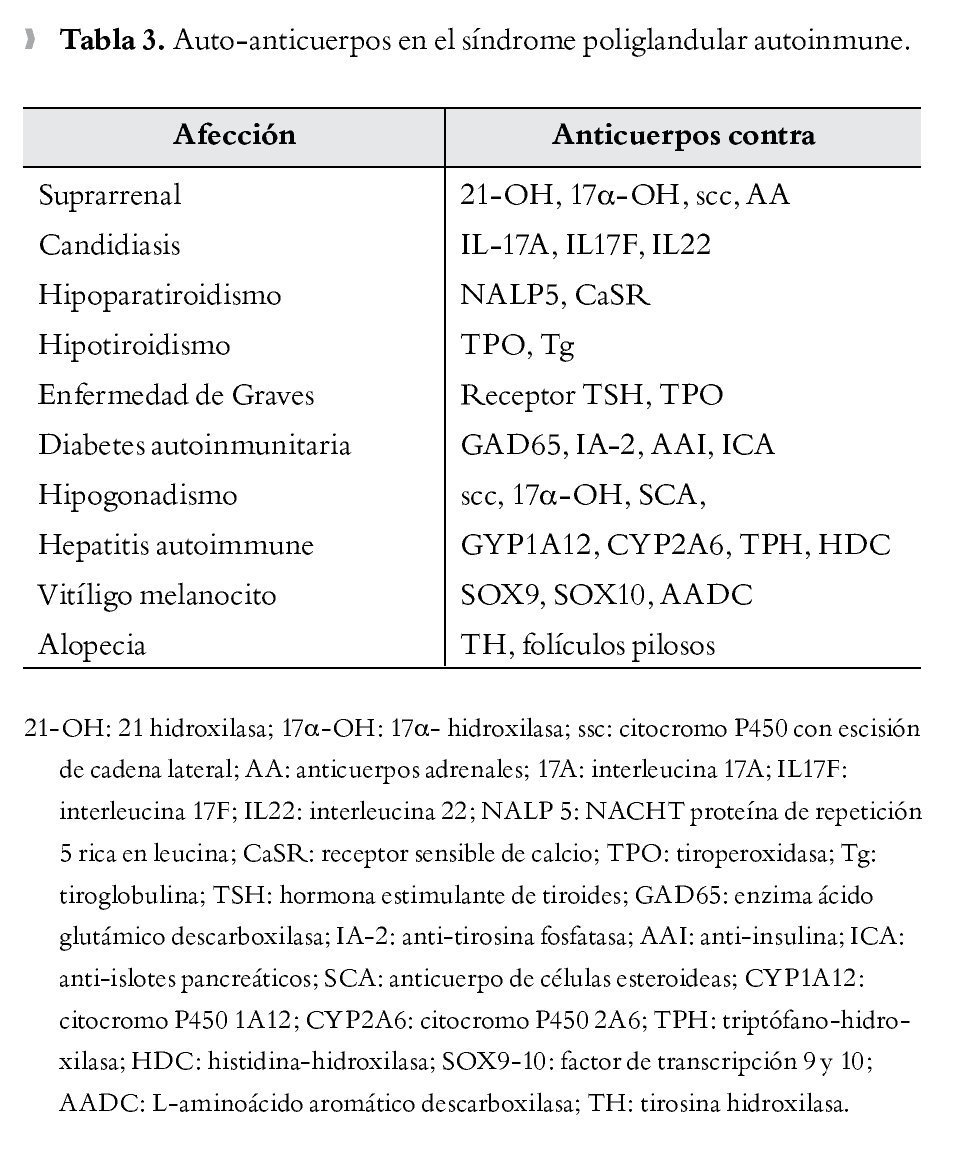
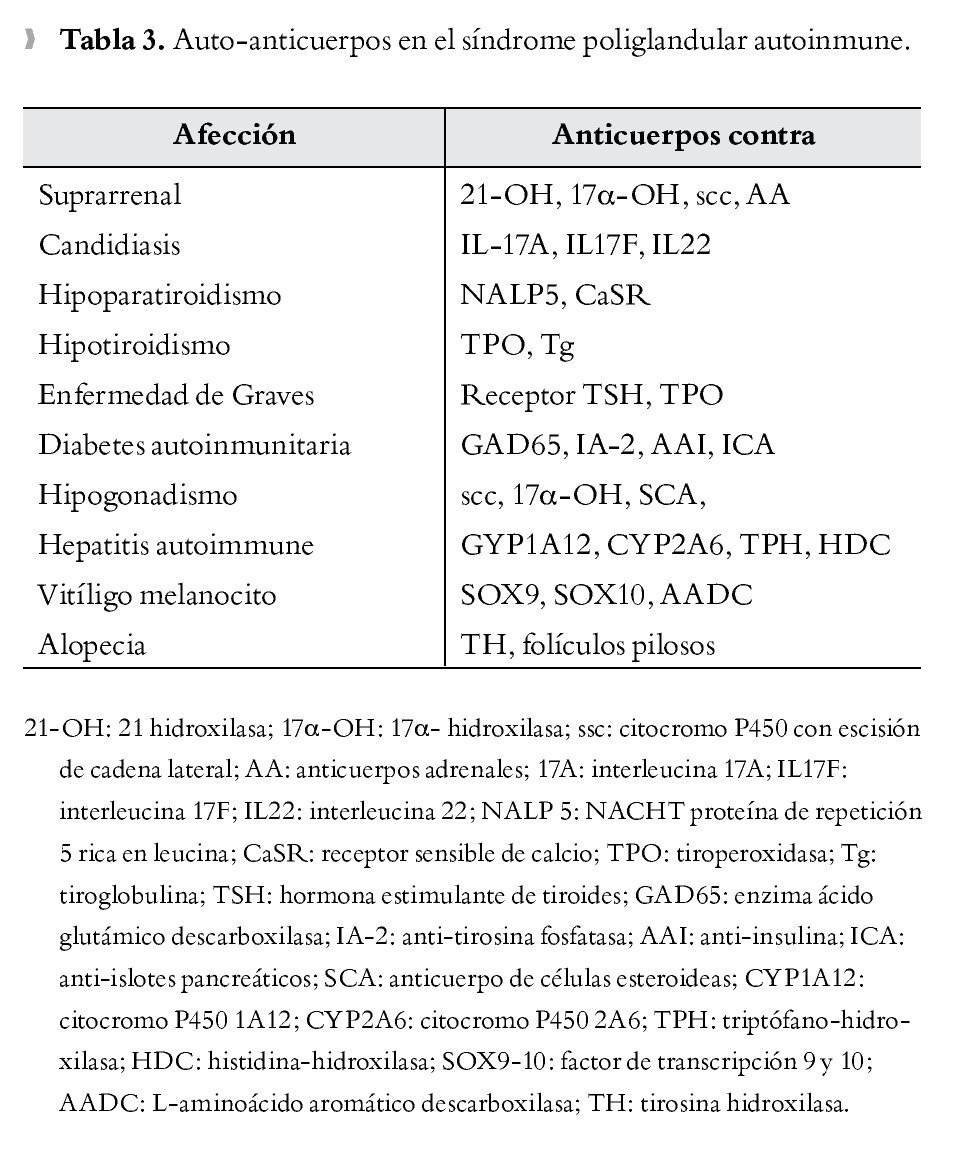
La afección está determinada por destrucción glandular mediada por células T, la cual es precedida por un periodo preclínico caracterizado por demostración de anticuerpos en sangre periférica que reaccionan contra antígenos específicos como proteínas, moléculas de superficie o enzimas intracelulares. Los pacientes con anticuerpos positivos (Tabla 3) se consideran en alto riesgo de desarrollar síntomas clínicos de hipofunción glandular, sin embargo se debe tener en cuenta que la determinación de anticuerpos no siempre conlleva el desarrollo ulterior de deficiencias glandulares.
Últimamente, numerosos investigadores 14,42,43 han propuesto los anticuerpos anti-interferón (Abs IFN-ω e Abs INF-α2) como parte del criterio diagnóstico del SPA tipo 1, dada la alta sensibilidad (86% a 98%)42,43 y especificidad (99%)43 para detectar mutaciones del gen AIRE, recomendándose la búsqueda de tales anticuerpos en pacientes jóvenes con presentaciones atípicas del síndrome tales como hepatitis inexplicable, queratopatía, rash asociado con fiebre, diarrea crónica o anemia perniciosa.42 Es importante señalar que dichos anticuerpos pueden encontrarse también presentes en pacientes con miastenia gravis y timoma. Se ha sugerido también la aparición de anticuerpos contra diversas citocinas, IL-17A, IL-17F, IL-22 como contribuyentes a la susceptibilidad por Cándida Albicans en el paciente con SPA tipo1.44
Anticuerpos contra enzimas esteroidogénicas como 21-hidroxilasa (21-OH), 17 a hidroxilasa (17-OH) y colesterol desmolasa (citP450scc) se encuentran frecuentemente en pacientes con ISR, los dos últimos junto con los anticuerpos contra células productoras de esteroides (anti-StCA) son también marcadores de insuficiencia gonadal en el contexto de un SPA.45 Se sabe que 21-OH es el principal autoantígeno identificado tanto en el SPA tipo 2 y en la ISR aislada,29 encontrándose también en 100% de pacientes con ISR del SPA tipo 1 en el grupo de Betterle.10 Entre los anticuerpos implicados en la predicción de hipoparatiroidismo se encuentran los dirigidos contra la NACHT leucine-rich-repeat protein 5 (NALP5, por sus siglas en inglés), expresada en el citoplasma de células principales paratiroideas, los cuales fueron encontrados en un 49% de los pacientes con SPA tipo 1 e hipoparatiroidismo46 y los anticuerpos contra el receptor sensible de calcio (CaSR) con menor trascendencia, puesto que su presencia parece depender del tipo de ensayo para determinarlo47 y sólo se presenta en una proporción pequeña de pacientes.25 La enfermedad tiroidea autoinmune es caracterizada por la presencia de anticuerpos antitiroglobulina (Tg) y antiperoxiasa (TPO), encontrándose en el 81% de los pacientes en el SPA tipo 2.35 Se ha visto que el tiempo entre la detección de anticuerpos y la presentación de la enfermedad tiroidea autoinmune es más largo que para otras entidades inmunomediadas.48 Por otra parte, la detección de autoanticuerpos tiroideos sin presencia de enfermedad tiroidea es encontrada en 27% de los pacientes con SPA tipo 1, manteniendo normal la función tiroidea durante el seguimiento.13
Para el desarrollo de DM1 se encuentran los anticuerpos contra la enzima ácido glutámico descarboxilasa (anti-GAD65), anticuerpos anti-insulina (IAA), los anticuerpos anti-islotes pancreáticos (ICA), y anti-tirosina fosfatasa (IA2), de estos los más frecuentes encontrados en el paciente con SPA tipo 1 son los GAD5, aunque esto no siempre se asocia a la aparición de DM1;49 en cambio, los IA2 muestran una alta especificidad sugiriendo ser el mayor predictor para el desarrollo de DM1.50 Se han encontrado anticuerpos contra célula parietal en pacientes con gastritis atrófica y anti factor intrínseco en pacientes con anemia perniciosa.49 Los anticuerpos dirigidos contra enzimas como la triptofanohidroxilasa (TPH) e histidina hidroxilasa (HDG) se han asociado a disfunción gastrointestinal en el contexto de SPA tipo 1, también han sido relacionado los anti-GAD65 dentro de dicha alteración.51 Principalmente anticuerpos contra enzimas del citocromo p450 como son 1A2 (PYP1A2) y 2A6 (CYP2A6), se ha encontrado en pacientes con hepatitis autoinmune.31 Pacientes con anticuerpos anti-microsomales de hígado-riñón (LKM-1), sin alteraciones en las pruebas de función hepática son identificados en el 25% de pacientes con SPA tipo 1.27 Se han encontrado también anticuerpos contra proteínas hipofisiarias, siendo identificado el TDRD6 como el mayor autoantígeno hipofisiario en el SPA tipo1.30 En este sentido los anticuerpos anti-pituitaria pueden ayudar a predecir la ocurrencia de hipopituitarismo, pero sólo cuando se considera el patrón de inmunotinción y los títulos de los mismos.52 En el vitíligo pueden existir anticuerpos contra tirosin-hidroxilasa, y factores de transcripción SOX9, SOX10.53,54 La presencia de autoanticuerpos sin evidencia de deficiencia glandular pueden preceder a la enfermedad, por lo tanto algunos autores recomiendan el seguimiento de estos pacientes en búsqueda de establecimiento de la enfermedad y repetir periódicamente los anticuerpos cada uno a dos años.27
• Tratamiento
El tratamiento del síndrome depende de las deficiencias glandulares encontradas, destacando que algunas de ellas son potencialmente letales si no son reconocidas y tratadas oportunamente. Por lo tanto, es pertinente hacer algunas aclaraciones: en los pacientes con ISR e hipoparatiroidismo concomitante durante el reemplazo esteroideo se puede presentar acentuación de la hipocalcemia, ya que la terapia esteroidea produce decremento en la absorción intestinal de calcio e incremento de la excreción renal del mismo,55 por lo que es importante tener en cuenta valores séricos de calcio para ajuste de tratamiento. También debe tenerse en cuenta que el reemplazo de la función tiroidea en el hipotiroidismo, debe realizarse descartando previamente ISR no tratada, ya que es conocida la posibilidad de precipitar crisis adrenal con la restitución de hormonas tiroideas por el aumento del aclaramiento hepático de cortisol.56
En el seguimiento de los pacientes con DM1, el requerimiento progresivo de dosis menores de insulina hace sospechar ISR concomitante.
La ISR se trata con reemplazo esteroideo, aumentándose la dosis en periodos de estrés. La dosis inicial recomendada de hidrocortisona 10 a 12.5 mg/ m2 por día,28 se prefiere dicho glucocorticoide por su vida media corta, la cual imita el ritmo circadiano normal del cortisol. Para evitar las complicaciones del uso crónico esteroideo se recomienda ajustar según el monitoreo de nivel de cortisol en orina, la dosis diaria a la más pequeña posible que controle la sintomatología clínica. La deficiencia de mineralocorticoide se realiza con acetato de fludrocortisona a dosis de 0.05 a 0.2 mg/día, usualmente en una sola dosis.28 La determinación de tensión arterial, renina plasmática y sobre todo potasio sérico, pueden servir de directrices para regular una adecuada dosis de medicamento. La infección crónica por Cándida albicans en el SPA tipo 1 generalmente es tratada a azoles: clotrimazol, ketoconazol, itraconazol y fluconazol, mención especial tiene el ketoconazol el cual puede interferir en la esteroidogénesis y provocar inhibición de la síntesis de cortisol. Se ha reportado incremento en la resistencia contra Cándida Albicans tras múltiples tratamientos.57 Los infantes con candidiasis mucocutánea deberán ser evaluados de manera periódica, en la búsqueda de anticuerpos que predigan la deficiencia glandular ulterior. El hipoparatiroidismo se trata con suplementos de calcio y vitamina, y por último los pacientes con anemia perniciosa y gastritis atrófica pueden cursar con grados variables de mala absorción, lo que interferiría con la absorción de calcio en un paciente con hipoparatiroidismo concomitante.
• Conflicto de intereses
Los autores declaran no tener conflicto de intereses.
• Financiamiento
No se recibió ningún patrocinio para realizar este artículo.
Correspondencia:
Av. Del Ferrocarril Nº 88 Esquina
Av. Indeco, Colonia Los Reyes Iztacala,
Tlalnepantla de Baz, Méx., México.
Teléfono: 2626 9200, ext. 2351.
Correo electrónico: drulises_navarrete83@hotmail.com