


We present the first case reported in the Republic of Colombia of associated trisomy 21-holoprosencephaly, one of the few in the world literature. The patient was a male newborn, the son of a healthy primiparous 19 year old. An obstetric sonogram at 27 weeks gestation revealed the foetus with both cerebral ventricles dilated, semilobar holoprosencephaly and cleft lip and palate. The mother received a detailed ultrasound scan and amniocentesis for foetal cytogenetic study. A caesarean section was performed at 38 weeks. The newborn weighed 2200g and was 46cm long. The head circumference was 28cm; thoracic girth, 28.5cm; and abdominal girth, 27cm. Apgar score was 6 at 1min, 6 at 5min and 9 at 10min. Physically, the newborn had a full moon face, mongoloid obliquity of the palpebral fissure, nasal bone hypoplasia, micrognathia and cleft lip and palate. Simple and contrast computed axial tomography of the brain showed semilobar holoprosencephaly and cleft lip. At 25h of life, the newborn expired from respiratory arrest.
Prenatal chromosome analysis presented a 47, XY, +21 G-band karyotype. Postnatal cytogenetic analysis, performed on umbilical cord blood using the fluorescent in situ hybridisation (FISH) technique with a locus specific identifier (LSI) 13/21 probe, showed the formula: nuc ish (D13ZX2), (D21ZX3) [30].
The cytogenetic aetiology of chromosome 21 and the holoprosencephaly gene are discussed, focusing on the fact that cytogenetic and gene alterations could function synergically and coincide in their expression with the postulate of the multiple-hit process.
Se presenta el primer caso reportado en la República de Colombia y otro de los pocos en la literatura médica mundial, de la asociación de trisomía 21 y holoprosencefalia. Paciente recién nacido, masculino, hijo de madre primípara sana de 19 años de edad. Con ultrasonido obstétrico realizado a las 27 semanas de gestación, se encontró feto con dilatación de ambos ventrículos cerebrales, holoprosencefalia semilobar, labio y paladar fisurados. A la madre se le realizó, ecografía de detalle y amniocentesis para estudio citogenético foetal. Se practicó cesárea a las 38 semanas, cuyo producto presentó peso de 2.200 g, talla de 46cm, perímetro cefálico de 28cm, perímetro torácico de 28,5cm y perímetro abdominal de 27cm. Puntuación de Apgar de 6 al primer minuto, 6 a los 5 min y 9 a los 10 min. Físicamente se observó cara de luna llena, oblicuidad mongoloide de las fisuras palpebrales, hipoplasia nasal, labio y paladar fisurados, micrognatia. La tomografía cerebral axial computarizada simple y con contraste mostró holoprosencefalia semilobar y labio fisurado. A las 25 h de vida, murió de paro respiratorio.
El cariotipo prenatal presentó fórmula cromosómica 47, XY, +21, por el método de bandas «G». El estudio citogenético posnatal, realizado con sangre de cordón umbilical y con el empleo de la técnica de FISH y la sonda LSI 13/21, mostró la fórmula: nuc ish (D13ZX2) (D21ZX3)[30].
Se discute la etiología citogenética del cromosoma 21 y la génica de la holoprosencefalia, pensando en el hecho de que alteraciones citogenéticas y génicas podrían trabajar de manera sinérgica y concordar en su expresión con el postulado del múltiple-hit process.
The diagnosis of this association confirmed using detailed ultrasound that makes it possible to visualise this central nervous system abnormality, with alteration in the early formation of the embryo prosencephalon; diagnosis is also confirmed by cytogenetic study, generally in amnioblasts, giving information as to the correct chromosome formula that the individual affected presents, but not about the degree of mental retardation or the capacity of learning. Because this syndrome is more frequent in young parents (due to the greater number of pregnancies presented in this age group), it would be of great interest to change the prenatal diagnosis protocol in the screening of young women at risk. Not only the routine genetic marker screening should be implemented, there should also be detailed ultrasounds between weeks 12 and 14 of gestation. This would make early detection of this type of association possible and allow for appropriate genetic counselling for the parents, in addition to better psychological preparation for receiving and handling children affected by these diseases.
IntroductionGiven the characteristics of the clinical phenotype of the case that we report, it is a good idea to present relevant information on trisomy 21 and its rare association with semilobar holoprosencephaly. Trisomy 21 is one of the most common genetic disorders. It was described for the first time in 1866, by John Langdon Haydon Down, who also called it “Mongolian idiocy”. Lejeune, in 1959, showed that this disorder was associated with an extra chromosome of the “G” group. Trisomy 21, or Down syndrome, is known to be phenotypically characterised by mental retardation and multiple malformations, within which cardiac anomalies and intestinal atresia are the most frequent. Other, less frequent malformations have been described, such as a flat occiput, brachycephaly, epicanthic folds, mongoloid palpebral fissures, Brushfield spots, strabismus and nystagmus, sunken nasal base, micrognathia, large fissured tongue, hypodontia, auricular hypoplasia with thickened helix, flat full-moon face with anterior–posterior flattening, short thick neck, excess nuchal fold tissue, diastasis of the rectus abdominis muscle, simian fold, fifth-finger clinodactyly and hypoplasia of the phalanges the same digit, ATD angle greater than 55°, radial loop on the ring finger, loop in the third interdigital space, radial loop in the hypothenar area, dysplastic pelvis with the iliac and acetabular angles lower than normal percentiles and cryptorchidism, and diastasis between the first and second knuckle. World prevalencia of Down syndrome is 1/800 births. However, the risk rises with the age of the mother, reaching 1/200 births when the mother is older than 35 years old.
This rare congenital malformation is the product of incomplete segmentation of the prosencephalon, Cohen (2006)1 mentioned that holoprosencephaly is a failure in the division of the embryonic structures of the anterior brain midline, which also caused defects in the middle of the face. De Myer et al. (1963)2 classified holoprosencephaly cases according to rising degree of severity as: lobar, semilobar and alobar. The most serious form is the alobar, in which the brain does not achieve separation and differentiation. It is associated with severe facial anomalies such as cyclopean deformity, lack of a nose, or cleft lip. In the semilobar type, the brain hemispheres tend to separate and it constitutes an intermediate form of the disease, while in the lobar type there is separation of the cerebral hemispheres and brain development can be almost normal. Dubourg et al. (2007)3 indicated that it presented in 1 of every 16,000 live births and that its aetiology is heterogeneous, including genetic factors (such as gestational diabetes) as well as chromosome factors (such as trisomy 13, 18 and 21).
Here we present a case of association of trisomy 21 and semilobar holoprosencephaly, the son of a 19-year-old mother and 20-year-old father, diagnosed prenatally through cytogenetics and detailed ultrasound scan.
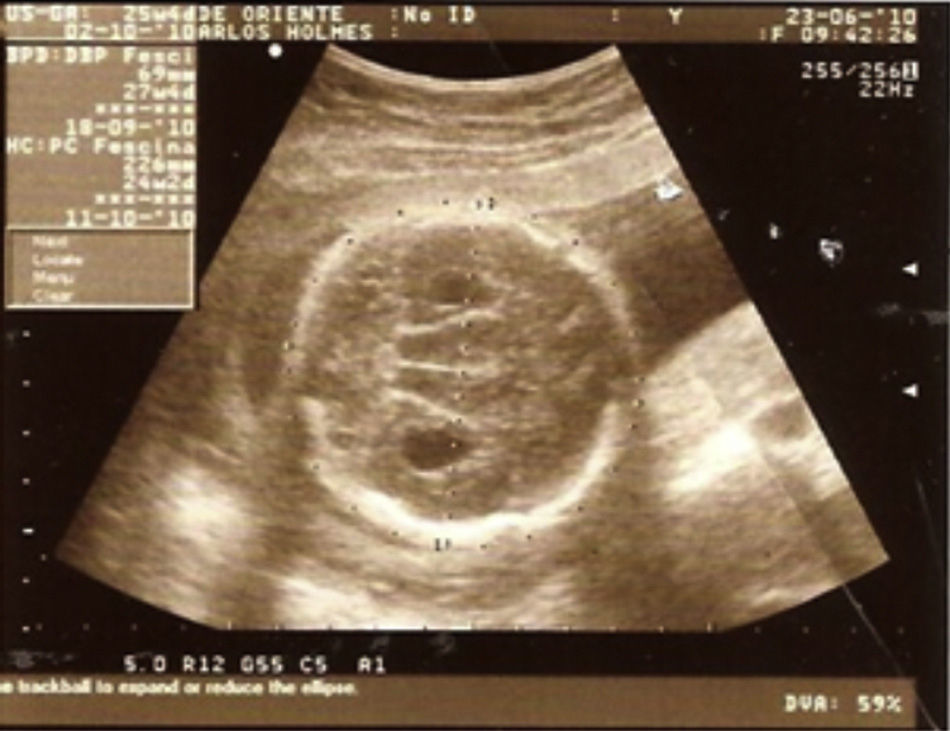
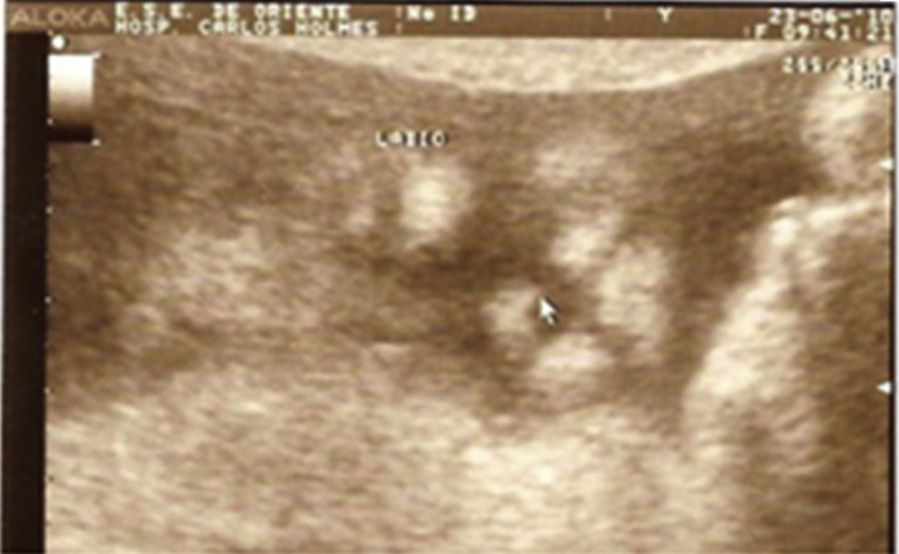
Case descriptionThe patient was a male newborn, the son of a healthy primiparous 19-year-old. An obstetric sonogram at 27 weeks of gestation revealed that the foetus had both cerebral ventricles dilated, semilobar holoprosencephaly and a cleft lip (Figs. 1 and 2).


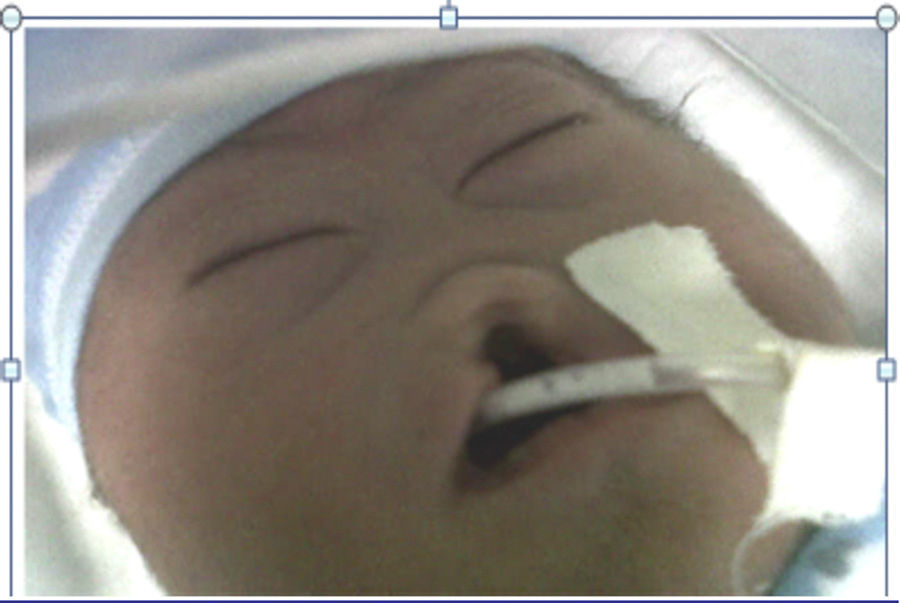
The mother was referred to the perinatology department, where she received a detailed ultrasound scan and amniocentesis to perform foetal cytogenetic study. A caesarean section was performed at 38 weeks. The newborn weighed 2200g and was 46cm in length. His head circumference was 28cm; thoracic girth, 28.5cm; and abdominal girth, 27cm. The Apgar score was 6 at 1min, 6 at 5min and 9 at 10min. The newborn was underweight for the gestational age and had mild respiratory distress, resolved with probe aspiration. No positive pressure ventilation was required. Physical examination revealed mongoloid obliquity of the palpebral fissures, nasal hypoplasia, a full-moon face, a single nasal fossa, and cleft plate and lip (Fig. 3). He required extra oxygen via hood and phenobarbital therapy to manage the convulsive syndrome. At 25h of life, the newborn went into cardiac arrest; he did not respond to resuscitation manoeuvres and expired.

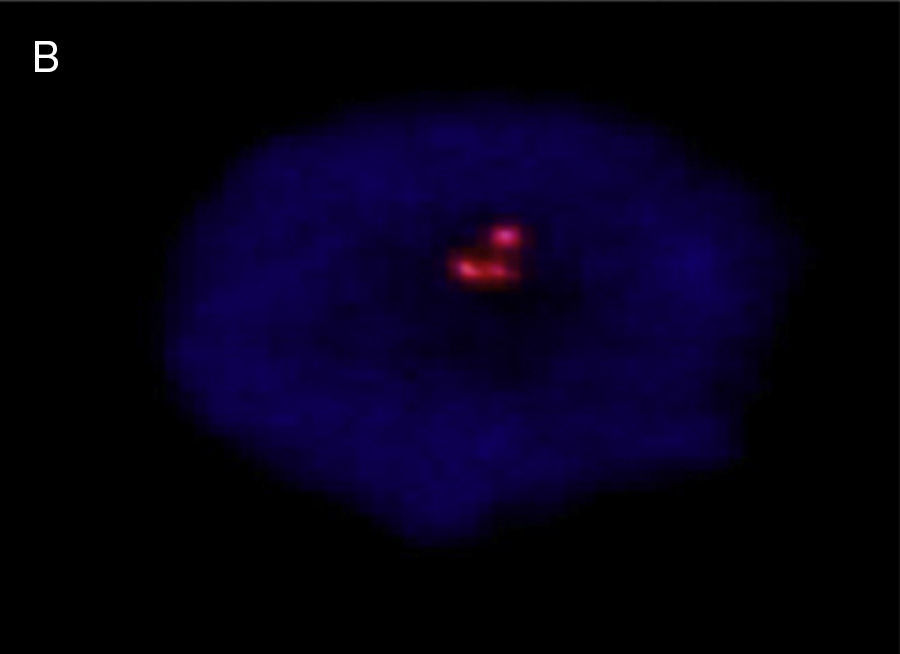
A prenatal cytogenetic study was performed using amniocentesis. Treated for G-bands, the results were a 47,XY,+21 karyotype. Umbilical chord blood was used for a postnatal cytogenetic study, using the fluorescence in situ hybridisation (FISH) technique, with CEP LSI 13/21 probes for “Y” and 21 chromosomes (ANEU VYSION DNA Probe), respectively; Carl Zeizz AXIOSCOP 40 and CytoLab View PathEx microscopes were utilised. The additional G group chromosome was ratified as 21 in interphase cell nuclei from the newborn (Fig. 4).
![Interphase nucleus – umbilical cord blood, showing 3 signals, with LSI 13/21 probes: nuc ish (D13ZX2) (D21ZX3) [30].](https://static.elsevier.es/multimedia/21719748/0000002000000002/v1_201608130021/S2171974815000264/v1_201608130021/en/main.assets/gr4.jpeg?xkr=ue/ImdikoIMrsJoerZ+w997EogCnBdOOD93cPFbanNcLh0A++iRI+X5zMTBgoGALlRaz965ds6MO9Xr/R18uHLB7ZQAibxomI5itoG1Kx2BmCXCQLtTrOg/1y3wyFGsQoULBDpRh1Em56cTceIxsLxW5oLEkb8SBbyNd8Kv1K4jQkUiaTzHHdCBs2ZKapcVYi833zIIVoq/1ee9Ol2f4mSpZcCtE8qR6eMngOvGCirCJRw3Attk+gbAF7BXtH4fEGuPthbas40vZnw9mBVai797ZQZvseZP5ao+BWlb45qQ= "Interphase nucleus – umbilical cord blood, showing 3 signals, with LSI 13/21 probes: nuc ish (D13ZX2) (D21ZX3) [30].")
The mother (who showed no important pathological, toxic or drug antecedents) also received a cytogenetic study, for which the necessary informed consent was given. A total of 12 metaphases revealed a modal number of 46 chromosomes (46,XX karyotype). The newborn's father was unavailable for cytogenetic study.
DiscussionOng et al. (1967)4 established that trisomy 21 was the most common human chromosomal aneuploidy and that it occurred with greater frequency associated with maternal meiotic non-disjunction of chromosome 21: 95% of the cases correspond to a free trisomy 21. Erdtmann (1981)5 indicated that there is an increase in constitutive heterochromatin variation in couples with meiotic errors, such as miscarriages and children with trisomy. Hussin et al. (2011)6 reported that, in non-disjunction, the effect of the recombination seemed to be located in the middle part of the chromosome arms, near the subtelomeric area.
Ming and Muenke (2002)7 consider holoprosencephaly to be etiologically heterogeneous. In addition, they believe that an individual's phenotype is the cumulative result of multiple influences, among which are environmental forces and genetic mutations, such as those reported for the Sonic hedgehog (SHH) (7q36), the first human gene described to produce it: ZIC2 (12q32.3); SIX3 (2p21); TGIF1 (18p11.31); PTCH (9q22.32); GLI2 (2q14.2); TDGF1 (3p21.31); DISP1 (1q41q42); FOXH1 (8q24.3); DLL1(6qter). These researchers have identified 4 main groups in the aetiology:
- 1.
Chromosome alterations of numbers 13, 18, 21 and structural alterations for chromosomes 21, 6 and 7.
- 2.
Cases with normal cytogenetics associated with gene problems, Smith–Lemil–Opitz syndrome; participation of 5%.
- 3.
Non-syndromic monogenic cases, in which there are generally heredo-familial antecedents of brain or facial disease, manifested discretely, whose inheritance pattern may be dominant autosomal, recessive or X-linked; between 4% and 9%.
- 4.
Environmental and teratogenic factors, as is the case of diabetes and ethylic ingestion.
The goal of this report was to raise the awareness of the increase in the number of the few cases published in world-wide medical literature, and of the first in our country (Colombia), of the association of trisomy 21 and holoprosencephaly resulting from the pregnancy of a young, healthy mother. It is necessary to think about the fact that cytogenetic and genetic alterations working synergistically could correspond in their expression with the hypothesis of the multiple-hit process outlined by Bendavid et al. (2009)8 and Rosenfeld et al. (2010).9
Conflict of interestsThe authors have no conflicts of interest to declare.