


Se presenta el primer caso reportado en la República de Colombia y uno de los pocos en la literatura médica mundial, de la asociación de trisomía 21 y holoprosencefalia. Paciente recién nacido, masculino, hijo de madre primípara sana de 19 años de edad. Con ultrasonido obstétrico realizado a las 27 semanas de gestación, se encontró feto con dilatación de ambos ventrículos cerebrales, holoprosencefalia semilobar, labio y paladar fisurados. A la madre se le realizó, ecografía de detalle y amniocentesis para estudio citogenético fetal. Se practicó cesárea a las 38 semanas, cuyo producto presentó peso de 2.200 g, talla de 46cm, perímetro cefálico de 28cm, perímetro torácico de 28,5cm y perímetro abdominal de 27cm. Puntuación de Apgar de 6 al primer minuto, 6 a los 5 min y 9 a los 10 min. Físicamente se observó cara de luna llena, oblicuidad mongoloide de las fisuras palpebrales, hipoplasia nasal, labio y paladar fisurados, micrognatia. La tomografía cerebral axial computarizada simple y con contraste mostró holoprosencefalia semilobar y labio fisurado. A las 25 h de vida, murió de paro respiratorio.
El cariotipo prenatal presentó fórmula cromosómica 47, XY, +21, por el método de bandas «G». El estudio citogenético posnatal, realizado con sangre de cordón umbilical y con el empleo de la técnica de FISH y la sonda LSI 13/21, mostró la fórmula: nuc ish (D13ZX2) (D21ZX3)[30].
Se discute la etiología citogenética del cromosoma 21 y la génica de la holoprosencefalia, pensando en el hecho de que alteraciones citogenéticas y génicas podrían trabajar de manera sinérgica y concordar en su expresión con el postulado del múltiple-hit process.
We present the first case reported in the Republic of Colombia of associated trisomy 21-holoprosencephaly, one of the few in the world literature. The patient was a male newborn, the son of a healthy primiparous 19 year old. An obstetric sonogram at 27 weeks gestation revealed the foetus with both cerebral ventricles dilated, semilobar holoprosencephaly and cleft lip and palate. The mother received a detailed ultrasound scan and amniocentesis for foetal cytogenetic study. A caesarean section was performed at 38 weeks. The newborn weighed 2200 g and was 46cm long. The head circumference was 28cm; thoracic girth, 28.5cm; and abdominal girth, 27cm. Apgar score was 6 at 1minute, 6 at 5minutes and 9 at 10minutes. Physically, the newborn had a full moon face, mongoloid obliquity of the palpebral fissure, nasal bone hypoplasia, micrognathia and cleft lip and palate. Simple and contrast computed axial tomography of the brain showed semilobar holoprosencephaly and cleft lip. At 25hours of life, the newborn expired from respiratory arrest.
Prenatal chromosome analysis presented a 47, XY, +21 G-band karyotype. Postnatal cytogenetic analysis, performed on umbilical cord blood using the fluorescent in situ hybridization (FISH) technique with a locus specific identifier (LSI) 13/21 probe, showed the formula: nuc ish (D13Zx2), (D21Zx3) [30].
The cytogenetic aetiology of chromosome 21 and the holoprosencephaly gene are discussed, focusing on the fact that cytogenetic and gene alterations could function synergically and coincide in their expression with the postulate of the multiple-hit process.
El diagnóstico de esta asociación se confirma con ecografía de detalle que permite visualizar esta anormalidad del sistema nervioso central, con alteración en la formación temprana del prosencéfalo del embrión y con un estudio citogenético, generalmente en amnioblastos, que informa de la fórmula cromosómica correcta que presenta el afectado, pero no del grado de retardo mental, o de su capacidad de aprendizaje. Dada la frecuencia de la trisomía 21 en hijos de parejas jóvenes, debido al mayor número de embarazos que se presentan en este grupo etario, sería de gran interés que, en su tamización en mujeres jóvenes en riesgo, en el diagnóstico prenatal se implementaran, en el protocolo correspondiente, no solo los marcadores genéticos de rutina, sino ecografía de detalle entre las semanas 12 y 14 de gestación, que permitan la detección precoz de este tipo de asociación, para proveer a los padres un asesoramiento genético adecuado, además de la mejor preparación psicológica, en la recepción y manejo de niños afectados con estos trastornos.
IntroducciónDadas las características del fenotipo clínico del caso motivo de esta comunicación, se presenta información relevante sobre la trisomía 21 y su inusual asociación con la holoprosencefalia semilobar. La trisomía 21 es uno de los trastornos genéticos más comunes. Fue descrito por primera vez en 1866 por John Langdon Haydon Down, quien también lo llamó «idiocia mongoloide». Lejeune, en 1959, demostró que este desorden estaba asociado con un cromosoma extra del grupo «G». Se conoce que la trisomía 21, o síndrome de Down, se caracteriza fenotípicamente por retardo mental y múltiples malformaciones, dentro de las cuales las anomalías cardiacas y atresia intestinal son las más frecuentes. Se han descrito otras menos frecuentes como occipucio aplanado, braquicefalia, pliegues epicánticos, hendiduras palpebrales mongoloides, manchas de Brushfield, estrabismo y nistagmo, base de la nariz deprimida, micrognatia, lengua grande y escrotal, hipodontia, hipoplasia auricular con hélix engrosado, cara aplanada, de luna llena con aplastamiento antero-posterior, cuello corto y ancho, exceso de pliegue epidérmico nucal, diastasis de rectos abdominales, pliegue simiano, clinodactilia del quinto dedo y pinzamiento de las falanges del mismo dedo, ángulo-atd mayor de 55°, presilla radial en el cuarto dedo, presilla en el tercer espacio interdigital, presilla radial en la región hipotenar, pelvis displásica con ángulo ilíaco y acetabular por debajo de los percentiles normales y criptorquidia, diastasis entre primer y segundo artejo. La prevalencia global del síndrome de Down es de 1/800 nacimientos, pero el riesgo se incrementa según la edad de la madre, llegando a ser mayor después de los 35 años, con 1/200 nacimientos.
En lo referente a la holoprosencefalia, poco frecuente, producto de una segmentación incompleta del prosencéfalo, Cohen (2006)1 menciona es una falla en la división de las estructuras embrionarias de la línea media en el cerebro anterior, que, además, causa defectos en la parte media de la cara. De Myer et al. (1963)2 clasificaron las holoprosencefalias según el grado de afectación creciente, en: lobar, semilobar y alobar. La alobar es la más grave, en la cual el cerebro no logra una separación y diferenciación. Se asocia con anomalías faciales graves como ciclopía, ausencia de nariz, o labio fisurado. En la semilobar, los hemisferios cerebrales tienen una tendencia a separarse y constituyen una forma intermedia de la enfermedad, mientras que en la lobar existe separación de los hemisferios cerebrales y el desarrollo del cerebro puede ser casi normal. Dubourg et al. (2007)3 mencionan que se presenta en uno de cada 16.000 nacidos vivos y su etiología es heterogénea, incluyendo factores genéticos como diabetes gestacional y cromosómicos como trisomía 13, 18 y 21.
Se presenta un caso de asociación de trisomía 21 y holoprosencefalia semilobar fruto de una madre y padre con 19 y 20 años de edad, respectivamente, diagnosticado prenatalmente por citogenética y ecografía de detalle.
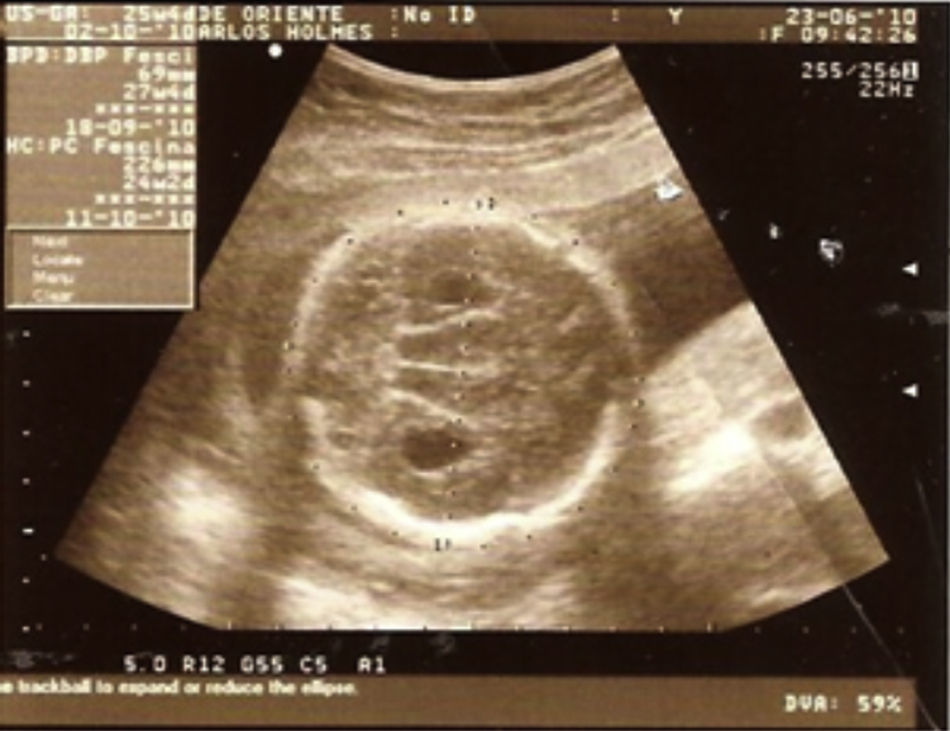
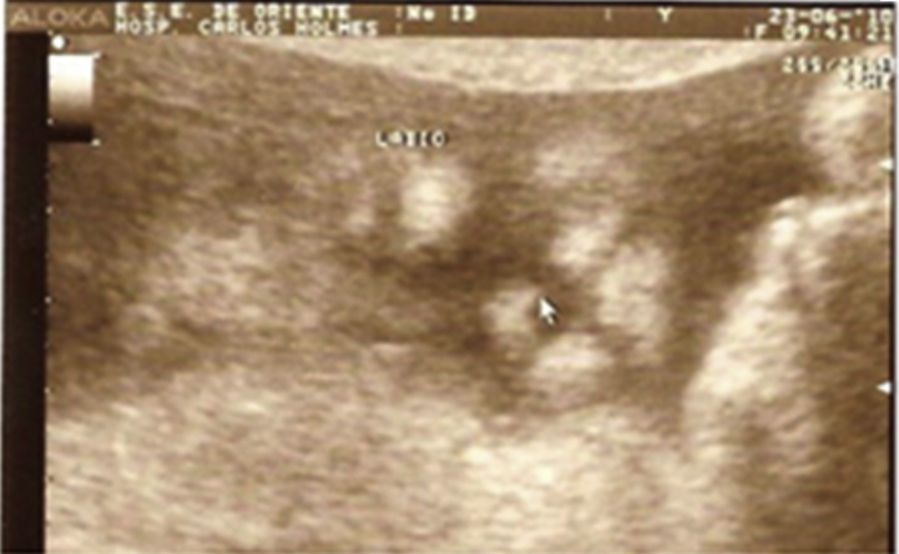
Descripción del casoPaciente masculino, producto del primer embarazo de una madre sana con 19 años de edad. Se realizó ultrasonido obstétrico a las 27 semanas, encontrándose dilatación de ambos ventrículos cerebrales, holoprosencefalia semilobar y labio fisurado (figs. 1 y 2).


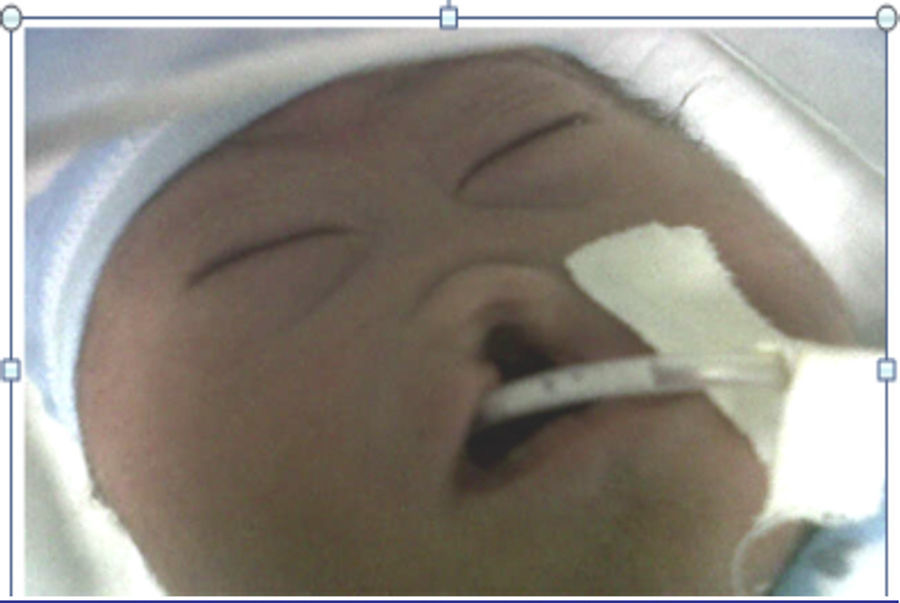
La paciente fue referida a perinatología donde le realizaron ecografía de detalle anatómico y amniocentesis para determinación de estudio citogenético fetal. Se practicó cesárea a las 38 semanas y se obtuvo un recién nacido de sexo masculino con peso de 2.200 g, talla de 46cm, perímetro cefálico de 28cm, perímetro torácico de 28,5cm y perímetro abdominal de 27cm. Puntuación de Apgar: 6 al primer minuto, 6 a los 5 min y 9 a los 10 min. Presentó además, peso bajo para la edad gestacional y dificultad respiratoria leve, que cedió a la aspiración con sonda. No requirió ventilación de presión positiva. Al examen físico se observó oblicuidad mongoloide de las fisuras palpebrales, hipoplasia nasal, cara de luna llena, fosa nasal única, labio y paladar fisurados (fig. 3). Requirió oxígeno complementario con cámara cefálica y terapéutica con fenobarbital para el manejo de un síndrome convulsivo. A las 25 h de vida, presentó paro cardiorrespiratorio, no respondió a maniobras de resucitación y falleció.

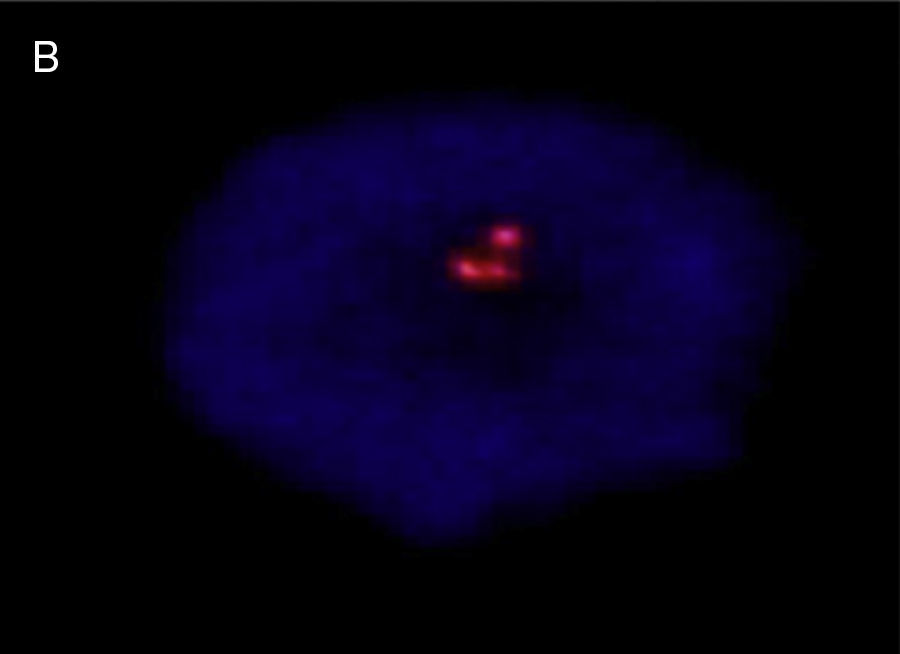
Los extendidos del estudio citogenético prenatal tras amniocentesis fueron tratados para bandas «G», cuyos resultados muestran fórmula cromosómica 47, XY, +21. También se realizó estudio citogenético posnatal en sangre de cordón umbilical con el empleo de la técnica de FISH, con el uso de las sondas CEP LSI 13/21 para cromosoma «Y» y cromosoma 21 (ANEU VYSION DNA Probe), respectivamente y el empleo de un microscopio Carl Zeizz AXIOSCOP 40 y del CytoLab View PathEx. El cromosoma adicional del grupo «G» fue ratificado como 21, en núcleos de células en interfase del paciente motivo de este reporte (fig. 4).
![Núcleo en interfase de sangre de cordón umbilical, que muestra 3 señales, con sondas LSI para cromosoma 13/21: nuc ish (D13ZX2)(D21ZX3)[30]](https://static.elsevier.es/multimedia/11382074/0000002000000002/v1_201608130039/S1138207415000172/v1_201608130039/es/main.assets/gr4.jpeg?xkr=ue/ImdikoIMrsJoerZ+w997EogCnBdOOD93cPFbanNcLh0A++iRI+X5zMTBgoGALlRaz965ds6MO9Xr/R18uHLB7ZQAibxomI5itoG1Kx2BmCXCQLtTrOg/1y3wyFGsQoULBDpRh1Em56cTceIxsLxW5oLEkb8SBbyNd8Kv1K4jQkUiaTzHHdCBs2ZKapcVYi833zIIVoq/1ee9Ol2f4mSpZcCtE8qR6eMngOvGCirCJRw3Attk+gbAF7BXtH4fEGuPthbas40vZnw9mBVai797ZQZvseZP5ao+BWlb45qQ= "Núcleo en interfase de sangre de cordón umbilical, que muestra 3 señales, con sondas LSI para cromosoma 13/21: nuc ish (D13ZX2)(D21ZX3)[30]")
Adicionalmente, a la madre, quien no mostró antecedentes patológicos, tóxicos ni farmacológicos importantes, y contando con el requisito de consentimiento informado, se le realizó estudio citogenético. Un total de 12 metafases mostraron un número modal de 46 cromosomas, fórmula 46, XX. El padre no estuvo disponible para el estudio citogenético.
DiscusiónOng et al. (1967)4 establecen que la trisomía 21 es la aneuploidía cromosómica humana más común y resulta con mayor frecuencia asociada a una no disjunción meiótica materna del cromosoma 21: el 95% de los casos corresponden a una trisomía 21 libre. Erdtmann (1981)5 menciona que, en parejas con errores meióticos, tales como abortos e hijos trisómicos, hay un aumento en las variantes con heterocromatina constitutiva. Hussin et al. (2011)6 comunican que en la no disjunción el efecto de la recombinación parece estar localizado en la parte media de los brazos cromosómicos y cerca de la región subtelomérica.
Ming y Muenke (2002)7 consideran la holoprosencefalia etiológicamente heterogénea y que el fenotipo de un individuo es un resultado acumulativo de múltiples influencias entre las cuales están las mutaciones génicas, como las reportadas para el Sonic hedgehog (SHH) (7q36), primer gen humano descrito en producirla: ZIC2 (12q32.3); SIX3 (2p21); TGIF1 (18p11.31); PTCH (9q22.32); GLI2 (2q14.2); TDGF1 (3p21.31); DISP1 (1q41q42); FOXH1 (8q24.3); DLL1(6qter), e influencias ambientales, adicionando que, en su etiología, se han identificado 4 grupos principales:
- 1.
Alteraciones cromosómicas de número 13, 18, 21 y de estructura para los cromosomas 21, 6 y 7.
- 2.
Casos con citogenética normal asociados con problemas génicos, síndrome de Smith Lemli Opitz, participación de un 5%.
- 3.
Casos no sindrómicos monogénicos, en donde generalmente hay antecedentes heredofamiliares de enfermedad cerebral, o facial, manifestada con discreción y cuyo patrón de herencia puede ser autosómico dominante, recesivo, o ligado al X, entre el 4 y el 9%.
- 4.
Factores ambientales y teratogénicos, como es el caso de la diabetes y la ingesta etílica.
El objetivo de este informe es el de poner en consideración el aumento del número de los pocos casos publicados en la literatura médica mundial y primero en nuestro país, Colombia, de la asociación de trisomía 21 y holoprosencefalia, resultante del embarazo de una madre sana y joven, pensando en el hecho de que alteraciones citogenéticas y génicas trabajando de manera sinérgica podrían concordar en su expresión con el postulado de múltiple-hit process, esbozado por Bendavid et al. (2009)8 y Rosenfeld et al. (2010)9.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.