



El síndrome de heterotaxia visceral o isomerismo visceral es el resultado de una falla en el establecimiento de la asimetría normal izquierda-derecha, durante el desarrollo embrionario. Los síndromes de heterotaxia visceral variedad asplenia y poliesplenia están presentes en el 3% de los pacientes con cardiopatía congénita, la incidencia de asplenia es de 1:20,000 nacidos vivos. Se presenta el siguiente caso de un recién nacido que fue valorado desde el periodo prenatal en el último trimestre de la gestación, el cual presentó un síndrome de isomerismo izquierdo sin interrupción del segmento hepático de vena cava inferior, con atrio único y 2 ventrículos, canal atrioventricular completo, sin atresia pulmonar y entrecruzamiento de ramas pulmonares, asociado a un bloqueo atrioventricular (AV) congénito, el cual representó un reto de abordaje diagnóstico y sobre todo terapéutico por la asociación de cardiopatía cianógena compleja de flujo pulmonar aumentado y bloqueo AV completo congénito.
Visceral heterotaxy syndrome or Visceral isomerism is the result of a failure in the establishment of left-right asymmetry normally during embryonic development. Visceral heterotaxy syndromes of asplenia and polysplenia variety are present in 3% of patients with congenital heart disease, the incidence of asplenia is 1:20.000 live births. We present the following case of a newborn who was assessed from the prenatal period in the last trimester of pregnancy, which presented a syndrome of left isomerism without interruption of hepatic segment of inferior vena cava, with single atrium and two ventricles, channel complete atrioventricular with pulmonary atresia and pulmonary branches intersecting associated with atrioventricular block (AV) congenital, which represented a challenge for diagnostic approach and therapeutic especially for complex cyanotic CHD association of increased pulmonary blood flow and congenital complete AV block.
Introducción
La heterotaxia visceral es el resultado de una falla en el establecimiento de la asimetría normal izquierda-derecha durante el desarrollo embrionario. Representa el 30% de los niños con malposición cardiaca, y el 45% de mortalidad atribuida a esta patología. Se ha estimado que de 4 a 10 niños nacidos vivos por 1,000 tienen una malformación cardiaca, 40% de los cuales son diagnosticados en el primer año de vida. Los síndromes de heterotaxia visceral variedad asplenia y poliesplenia están presentes en el 3% de los pacientes con cardiopatía congénita, la incidencia de asplenia es de 1:20,000 nacidos vivos1-5.
Presentación del caso
Se presenta el caso clínico de una recién nacida femenina, producto de la gesta 1 de madre de 20 años de edad aparentemente sana, de embarazo normoevolutivo, con control prenatal irregular desde el 4° mes de gestación, con 4 consultas en total, se niegan amenazas de aborto y de parto pretérmino, se realizaron 3 ultrasonidos (US) obstétricos, reportando en medio privado una probable anomalía de ebstein. Acude a valoración por Medicina Materno-fetal y Cardiopediatría de nuestro instituto, diagnosticando un canal AV con cabalgamiento aórtico del 50%, sin lograr observar vaso pulmonar. Se obtiene de término vía abdominal, con un peso al nacer de 2,498 g, talla 45 cm, Apgar 8-9, perímetro cefálico 34 cm, perímetro torácico 31 cm, perímetro abdominal 28 cm, frecuencia cardiaca 99x´, frecuencia respiratoria 35x´, saturación 98%. Se mantuvo en la Unidad de Cuidados intensivos neonatales, presentó bradicardia a partir del 2° día de vida, desaturación a partir del 3° día de vida entre 80% y 85%, por lo cual se dejó con oxígeno a flujo indirecto inicialmente y posteriormente, al 4º día de vida se decide su intubación orotraqueal. Fue valorado por Cardiopediatría al primer día de vida, estableciendo los siguientes hallazgos ecocardiográficos: situs ambiguo, yuxtaposición aorto-cava a la izquierda, levocardia, retornos venosos sistémicos y pulmonares normales, cavidad atrial única, con escaso remanente superior del septum interatrial, válvula atrioventricular única, con insuficiencia leve de su porción izquierda, con inserciones en la pared libre de ambos ventrículos, con un diámetro de 15 mm, defecto septal interventricular en su porción de entrada, con cabalgamiento aórtico mayor del 50%, desplazamiento anterior y a la derecha del septum infundibular, anillo aórtico de 5 mm (Z -3), anillo pulmonar de 10 mm (Z +4), tronco de la arteria pulmonar 10 mm (Z +4), se observa entrecruzamiento de ramas pulmonares, rama derecha 4 mm (Z +1), rama izquierda 4.5 mm (Z +2), arco aórtico izquierdo sin obstrucción. Se estableció diagnóstico de heterotaxia visceral variedad poliesplenia (fig. 1). El paciente requería para su corrección una cirugía de tipo univentricular tipo Damus Kaye Stensen modificada, la cual consiste en garantizar el flujo sistémico al seccionar el tronco pulmonar y anastomosarlo a la porción ascendente de la aorta, colocar una fístula sistémico pulmonar tipo Blalock Taussig modificado, y colocar un marcapaso definitivo, todo ello, conlleva una alta mortalidad operatoria, es por esto que requería atención en 3° nivel.
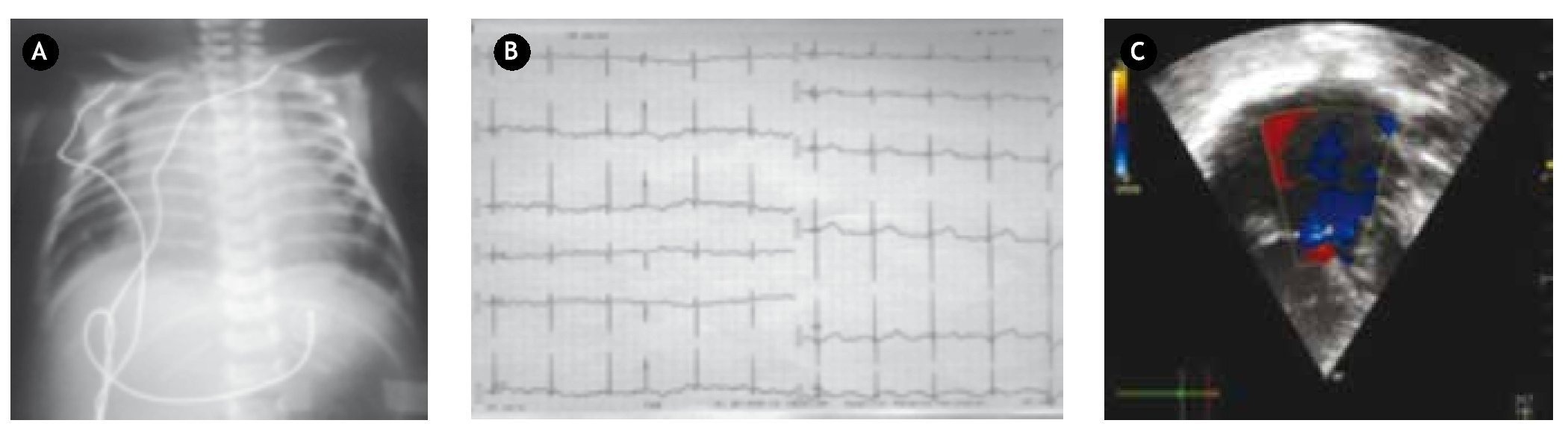
Figura 1. A) Radiografía de tórax muestra situs ambiguo, hígado central, cardiomegalia grado IV, flujo pulmonar aumentado. B) Electrocardiograma muestra bloqueo AV completo, con una frecuencia ventricular de 65x' y una frecuencia atrial de 115x´, eje P +150°, eje QRS +130°, ondas R puras en V1 y V6, patrón RS de V2 a V5. C) Eje apical 4 cámaras en el que se observa una cavidad atrial única, con válvula atrioventricular única, 2 ventrículos con un defecto septal interventricular amplio, con Doppler color se observa doble jet regurgitante de la válvula AV única.
Se mantuvo con manejo a base de furosemida, espironolactona y captopril, se agregó dobutamina, mantuvo frecuencia cardiaca entre 65-70x' a pesar de manejo, se diagnosticó un bloqueo atrioventricular completo. Se solicitó su traslado a 3° nivel de atención, sin lograr su traslado, presentando paro cardiorrespiratorio a los 13 días de vida, sin revertir a maniobras avanzadas de reanimación.
Discusión
Este tipo de síndromes representan un reto diagnóstico y terapéutico para un Servicio de Medicina Materno Fetal, así como para el servicio de Cardiología Pediátrica y de Cirugía Cardiovascular, ya que se requiere de toda una infraestructura desde el punto de vista cardiovascular para ofrecer de manera oportuna el tratamiento quirúrgico, hemodinámico y electrofisiológico adecuado para este tipo de pacientes con el fin de ofrecer una mejor sobrevida.
Nuestro caso tiene la particularidad de que no presentó interrupción del segmento hepático de vena cava inferior, hallazgo característico (95%) en este tipo de síndromes de isomerismo izquierdo, más no determinante para clasificar este tipo de cuadros sindrómáticos, sin embargo, se requeriría de un estudio angiográfico para demostrar este hallazgo, por otro lado, si se relacionó en el periodo neonatal con la presentación de un bloqueo atrioventricular congénito que fue determinante para el cuadro de defunción del paciente. Por tratarse de una cardiopatía de flujo pulmonar aumentado, requirió de manejo con diúreticos de asa e inhibidores de la enzima convertidora de angiotensina.
Se intentó su traslado a 3er nivel de atención ya que el paciente requería para su corrección una cirugía de tipo univentricular tipo Damus Kaye Stensen modificada la cual consiste en garantizar el flujo sistémico al seccionar el tronco pulmonar y anastomosarlo a la porción ascendente de la aorta, colocar una fístula sistémico pulmonar tipo Blalock Taussig modificado, y finalmente colocar un marcapaso definitivo, todo ello conlleva una alta mortalidad operatoria, dado que no contamos con este tipo de servicio e infraestructura en nuestro hospital, se intentó su traslado.
Conclusión
Los síndromes de heterotaxia visceral son enfermedades raras pero diagnosticables en el período neonatal como este caso, que requieren de una atención de alta especialidad por parte del personal que asume la responsabilidad de atender estos casos, asi como de una infraestructura adecuada desde el punto de vista cardiovascular que pueda resolver de manera oportuna y con éxito en la sobrevida y morbilidad de estos pacientes tan complejos.
Asimismo, se debe insistir que la detección prenatal de este tipo de casos es fundamental que se realice en las primeras 18 a 22 semanas de gestación con el fin de establecer las estrategias terapéuticas necesarias para decidir continuar con la gestación o sugerir la terminación de la misma en base a un comité de ética que oriente a los padres exponiendo el tipo de complejo sindromático y el pronóstico de sobrevida del paciente.
Financiamiento
No se recibió ningún patrocinio para llevar a cabo este artículo.
Conflicto de intereses
El autor declara no tener conflicto de intereses.
* Autor para correspondencia:
jodolphin79@hotmail.com (J. O. Osorio-Díaz).