


El estudio de la estructura, fisiología y trastornos relacionados con la mitocondria han sido temas de investigación para muchos científicos a lo largo de la historia, podemos remitirnos al año de 1951 en el que Denis Leigh describe el caso de un niño con las manifestaciones clínicas propias del síndrome que hoy en día lleva su nombre, del cual cada vez se conoce más pero se avanza poco en el establecimiento de un tratamiento que logre elevar la esperanza de vida de quienes lo padecen.
El cuerpo humano como máquina perfecta requiere del aporte de combustibles biológicos (carbohidratos, lípidos y proteínas) necesarios para obtener la energía química que requieren diversos procesos biológicos y de esta manera mantener la homeostasis. El metabolismo de estos combustibles en el organismo se logra a través del uso de distintas vías catabólicas y anabólicas, las cuales requieren del coordinado funcionamiento de elementos celulares, es decir de la buena comunicación entre los orgánulos de cualquier célula en el organismo.
Tras el correcto metabolismo de los combustibles biológicos en las células, finalmente obtenemos moléculas de energía química en forma de ATP que son necesarias para llevar a cabo procesos tales como; paso de moléculas a través de la membrana celular, movimiento de flagelos, contracción muscular, entre muchas otras funciones vitales. La mayor parte de esta energía química se obtiene gracias a la participación de la mitocondria, en cuya estructura se localiza la cadena respiratoria importante para llevar a cabo la fosforilación oxidativa y finalmente la obtención de ATP.
Identificación conceptual del síndrome de LeighTrastorno neurodegenerativo secundario a la ausencia congénita de: Complejo Piruvato Deshidrogenasa, Complejo I, IV o V de la cadena respiratoria1.
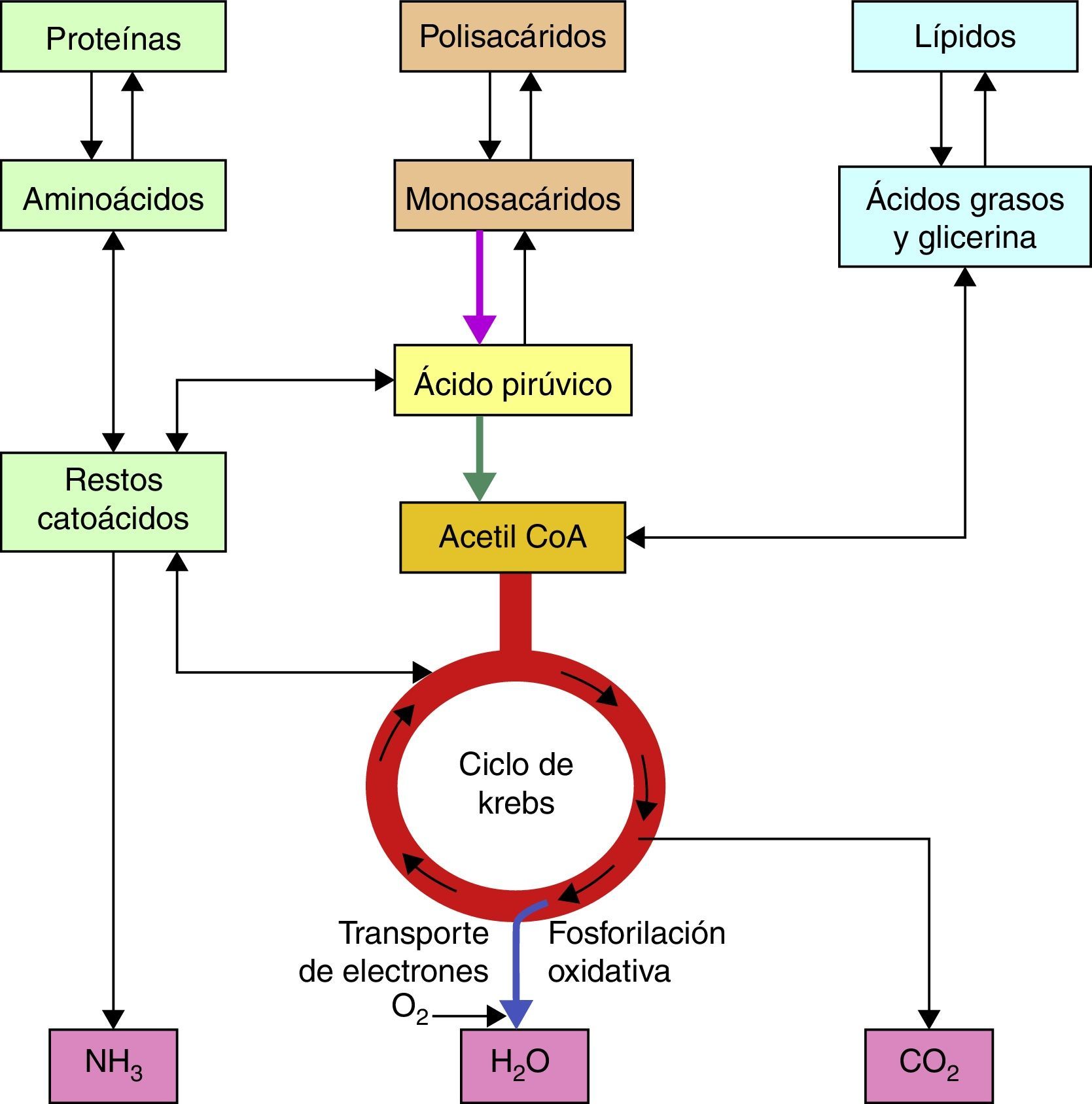
Bases bioquímicas del síndrome de LeighLas vías metabólicas que se utilizan tanto en el catabolismo de carbohidratos, lípidos y proteínas convergen en la mitocondria tras la formación final de moléculas de Acetil CoA2, la cual será metabolizada posteriormente por el ciclo de los ácidos tricarboxílicos para obtener cofactores reducidos, NADH + H y FADH2 que en seguida serán oxidados en la cadena respiratoria para la obtención de las moléculas de ATP (fig. 1).

En el caso de los carbohidratos, tanto la glucólisis aeróbica como anaeróbica son vías importantes para el aprovechamiento de estas moléculas, tras la serie de reacciones que implica la glucólisis anaeróbica en el citosol celular, llegamos a la obtención de Piruvato, el cual será sustrato para el complejo piruvato deshidrogenasa quien oxidará la molécula de piruvato en Acetil CoA en la mitocondria.
La molécula de Acetil CoA se forma a partir de Coenzima A, cuya molécula contiene adenina, ribosa, ácido pantoténico y un grupo tiol, este último reacciona con los grupos carboxilo para que finalmente se obtenga la molécula de Acetil CoA, es un compuesto de alta energía por lo que puede donar su grupo acetilo en el ciclo de los ácidos tricarboxílicos.
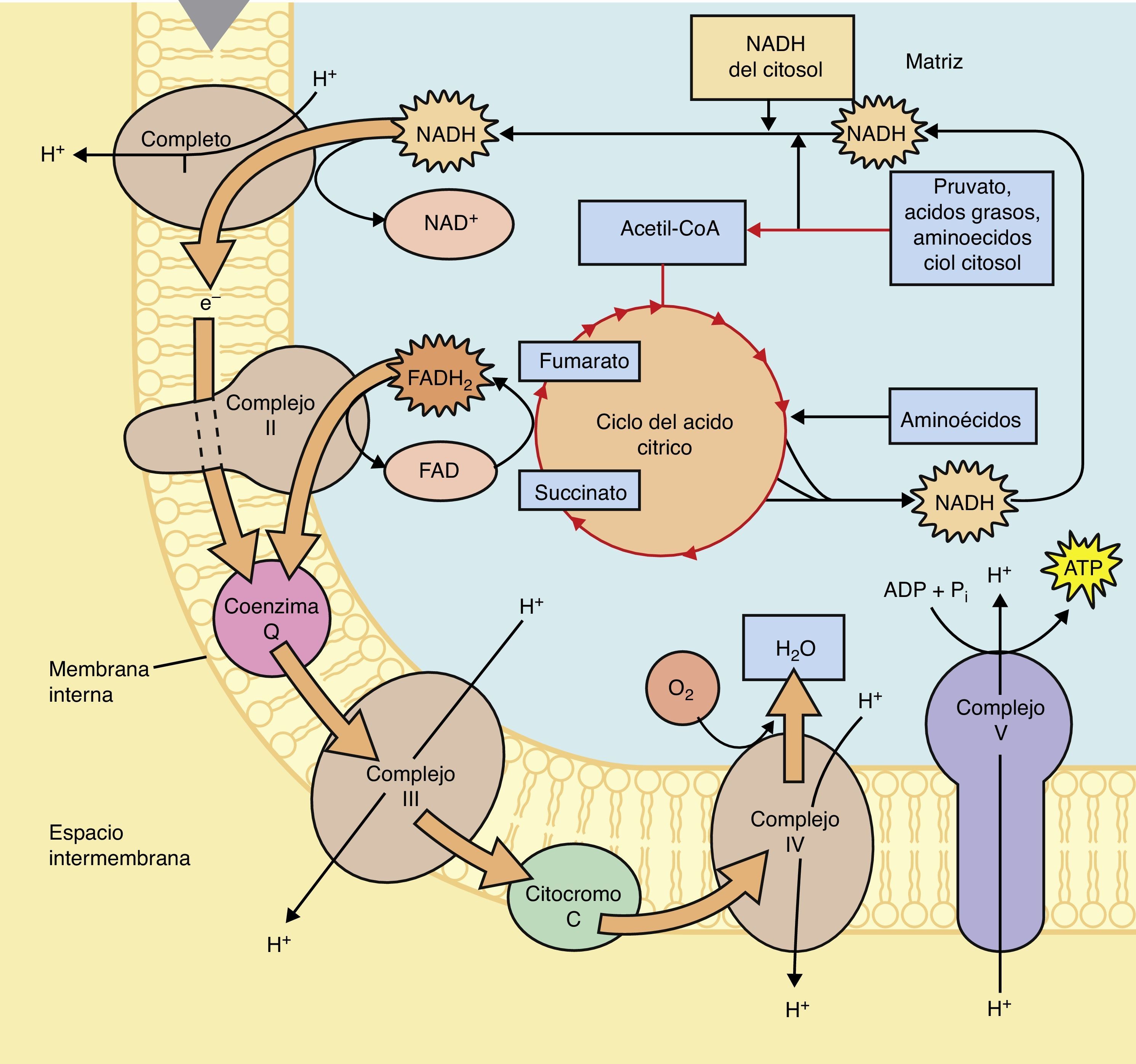
En el ciclo de los ácidos tricarboxílicos la molécula de Acetil CoA será metabolizada hasta CO2 y H2O, así mismo en la serie de reacciones que forman parte de este ciclo se obtienen; 3 NADH + H, 1 FADH2 y una molécula de GTP. Los cofactores reducidos (NADH + H y FADH2) que se producen en este ciclo son importantes debido a que fungen como transportadores de los electrones que se obtienen tras el catabolismo de los combustibles biológicos, por tanto, estos cofactores resultantes del ciclo de Krebs o de los ácidos tricarboxílicos, donarán posteriormente dichos electrones al primer complejo de la cadena respiratoria en el caso del cofactor NADH + H o al Complejo II en el caso del cofactor FADH2, tras una serie de reacciones en el que estos cofactores se oxidan y los complejos se reducen (fig. 2).

Los cofactores reducidos que se obtienen en la glucólisis anaeróbica del citosol celular no pueden atravesar la membrana mitocondrial, por lo tanto, para ingresar hacen uso de las lanzaderas glicerol – 3 – fosfato (principalmente en células cerebrales y musculares) y malato – aspartato (principalmente en hígado y corazón), las cuales introducen los electrones de los cofactores reducidos desde el citoplasma hacía la mitocondria.
El paso de electrones a través de la cadena respiratoria comienza en el complejo I o II, posteriormente pasan a la ubiquinona (Q), en seguida al complejo III, este reduce al citocromo y finalmente el citocromo reduce al complejo IV, el cual donará 2 electrones al Oxigeno para la formación de una molécula de Agua, cabe destacar que el paso de los electrones a través de los complejos enzimáticos de la cadena respiratoria se lleva a cabo desde el complejo que presenta un Potencial de Reducción Estándar menor hacía el complejo que tiene un Potencial de Reducción Estándar mayor, es decir, desde el complejo con una menor afinidad por los electrones hacía el complejo que tiene una mayor afinidad por los mismos.
El paso de los electrones a través de los complejos de la cadena respiratoria está acoplado al bombeo de protones por parte del complejo I, III y IV hacía el espacio intermembranal, creando de esta forma un gradiente electroquímico en ese espacio, la tendencia de los protones para regresar hacía la matriz y estabilizar el gradiente a ambos lados de la membrana mitocondrial interna va a permitir que pasen a través del complejo ATPasa, cuya estructura se basa en una subunidad F1 y F0. La subunidad F0 forma el canal a través del cual pasan los protones, mientras que la subunidad F1, es el sitio en el cual, a partir de ADP y Pi, se lleva a cabo la formación de la molécula de ATP, por tanto es importante destacar que la energía que se genera tras el paso de los protones por este canal permite que se libere la molécula de ATP que se ha formado por los sustratos (ADP y Pi), no se debe pensar que la energía que se obtiene por el paso de los protones es propiamente la energía que contiene el ATP.
Tras la obtención de moléculas de ATP, estas pueden ser utilizadas en la gran gama de procesos fisiológicos que lo requieren para un buen funcionamiento y el mantenimiento de un equilibrio vital para el ser humano.
FisiopatologíaEn el organismo hay tejidos que destacan por el requerimiento de un metabolismo oxidativo intacto que asegure la obtención de la energía necesaria para llevar a cabo de manera eficaz su función, dentro de estos tejidos es preponderante el sistema nervioso central, dentro del cual los ganglios basales destacan por presentar una elevada actividad metabólica, situación que los hace vulnerables a los daños celulares, por lo que fallas en el metabolismo celular de estas estructuras se verá reflejado de manera significativa en las funciones de control que llevan a cabo en el organismo.
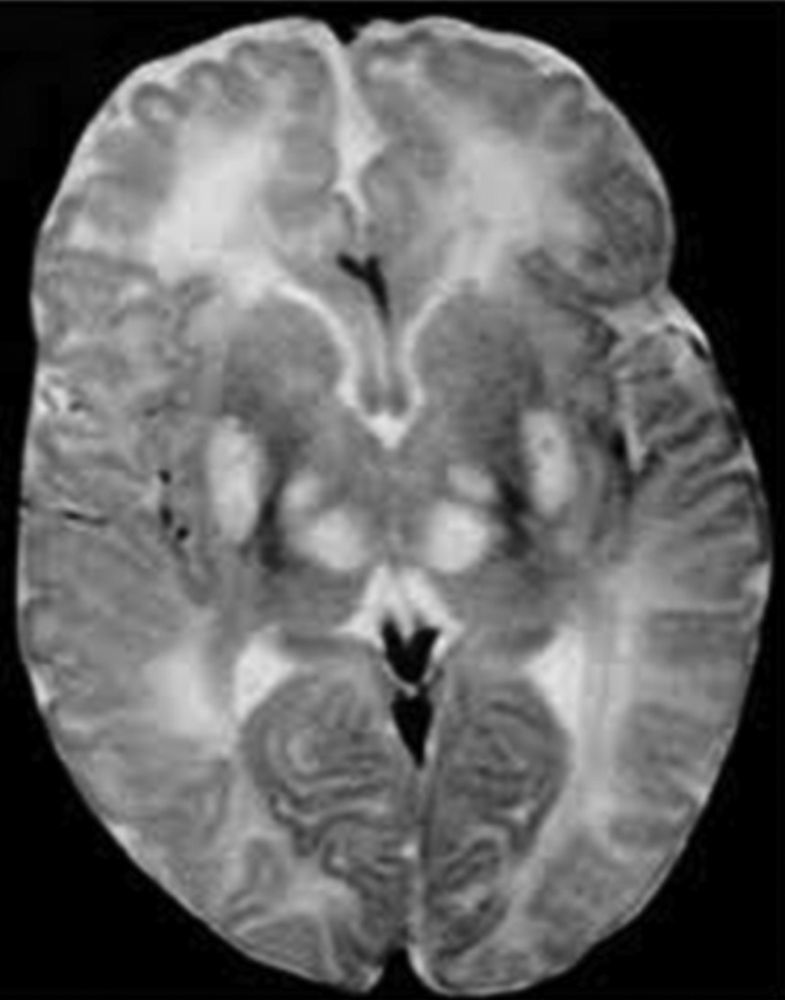
Los ganglios basales (globo pálido, putamen, núcleo caudado, núcleo subtalámico y sustancia negra) son acumulaciones de cuerpos neuronales que tienen como función primordial el control del inicio, amplitud y rapidez de los movimientos4, en un niño con síndrome Leigh la falla en el metabolismo oxidativo y la necrosis de estas estructuras se traduce en defectos del movimiento, por tanto se caracterizan por déficit motor o activación anormal del sistema motor, lo que da lugar a rigidez, temblor y movimientos involuntarios (fig. 3).

La disminución en la producción de ATP que se presenta en este síndrome no se limita en sus afectaciones al Sistema Nervioso Central, las consecuencias de esta disminución de energía se ven reflejadas en las manifestaciones clínicas propias del Síndrome de Leigh.
Manifestaciones clínicasCrisis convulsivas, Retraso psicomotor, Atrofia óptica, Hipotonía, Debilidad, Letargo, Vómitos, Movimientos anormales (ataxia, temblor), Irritabilidad, Pérdida de visión y Anormalidades respiratorias3.
Herencia del síndrome de LeighA pesar de que la mitocondria cuenta con su propio ADN, no es independiente de la participación del ADN nuclear, ya que ambos participan en la codificación de las subunidades que conforman cada uno de los complejos enzimáticos de la cadena respiratoria, a excepción del Complejo II que está exclusivamente codificado por el ADN nuclear, es por eso que dependiendo del complejo afectado y de su codificación, el tipo de herencia puede ser: Materna, en el caso de mutaciones en ADN mitocondrial o Autosómica Recesiva, cuando las mutaciones se dan en el ADN nuclear5.
Los casos más reportados de síndrome de Leigh han sido identificados por mutaciones en el ADN mitocondrial, cuyo gen codifica para una de las subunidades de la ATPasa, por lo que presenta una herencia materna, por otro lado, mutaciones en el ADN nuclear que afectan al complejo IV el síndrome presenta herencia Autosómica Recesiva. La frecuencia de presentación de este síndrome es de 1 de cada 36,000 nacimientos.
Diagnóstico y tratamientoLas manifestaciones clínicas del síndrome se presentan antes de los 6 meses de edad, el médico que evalúa el caso requiere de una gran destreza para poder diferenciar este trastorno de otros trastornos mitocondriales que presentan características similares y con los cuales se puede confundir, para un diagnóstico diferencial adecuado se debe hacer uso de recursos como; biopsia muscular para analizar el funcionamiento de la cadena respiratoria y poder identificar el error en los complejos enzimáticos, así mismo el uso de resonancia magnética y tomografía computarizada que permite identificar la necrosis de los ganglios basales, característica principal del síndrome de Leigh.
El tratamiento tras el diagnóstico de este síndrome es paliativo, implementar una dieta cetogénica cuando el síndrome se asocia a deficiencia de Piruvato Deshidrogenasa puede no reflejar ningún beneficio, debido a la falta de un tratamiento eficaz que permita reducir los daños, la esperanza de vida de un niño con este síndrome no supera los 2 años.
FinanciamientoNo se recibió patrocinio para llevar a cabo este artículo.
Conflicto de interesesEl autor declara no tener conflicto de intereses.
Al Dr. Aurelio Mendoza Medellin, catedrático de la unidad de aprendizaje de Bioquímica de la Facultad de Medicina de la Universidad Autónoma del Estado de México, por los conocimientos aportados que influyeron para llevar a cabo este artículo.