


Torpedo maculopathy is a rare oval-shaped hypopigmented lesion in the temporal part of the macula. Its main feature is its fusiform appearance. It is not usually associated with any decrease in visual acuity and remains stable over time.
We report the case of a girl aged four years with torpedo maculopathy and hypo-autofluorescence in the right eye. In contrast, autofluorescence was normal in the only previous case reported in which autofluorescence was described in such a young patient.
La maculopatía en torpedo es una rara lesión oval hipopigmentada localizada en la zona temporal de la mácula cuya principal característica es su morfología fusiforme. No suele asociar disminución en la agudeza visual y permanece estable en el tiempo.
Presentamos el caso de una niña de 4 años con maculopatía en torpedo en su ojo derecho que se mostró hipoautofluorescente en contraste con el único caso en la bibliografía en el que se describe la autofluorescencia de una paciente tan joven, en el que la autofluorescencia fue normal.
Torpedo maculopathy (TM) is a rare, benign condition that affects the retinal pigment epithelium (RPE) and has a characteristic shape and location. It is usually diagnosed incidentally in asymptomatic patients1–4 and is normally unilateral, although bilateral cases have been reported.5 In 1992 it was described by Roseman and Gass6 as a hypopigmented nevus of the RPE. These authors considered it a type of nevus in which the RPE cells were flattened and not enlarged. The term TM was first used by Daily in 1993 and it has become generally adopted.7
Only isolated case reports or series with few cases have been reported, and so there are no reliable epidemiological data regarding race, gender or other demographic data.8–10
We present the OCT and autofluorescence findings for a child with TM.
Case studyGirl aged 4 years, no relevant clinical history, referred for a routine eye examination. Best-corrected visual acuity was 20/20 bilaterally.
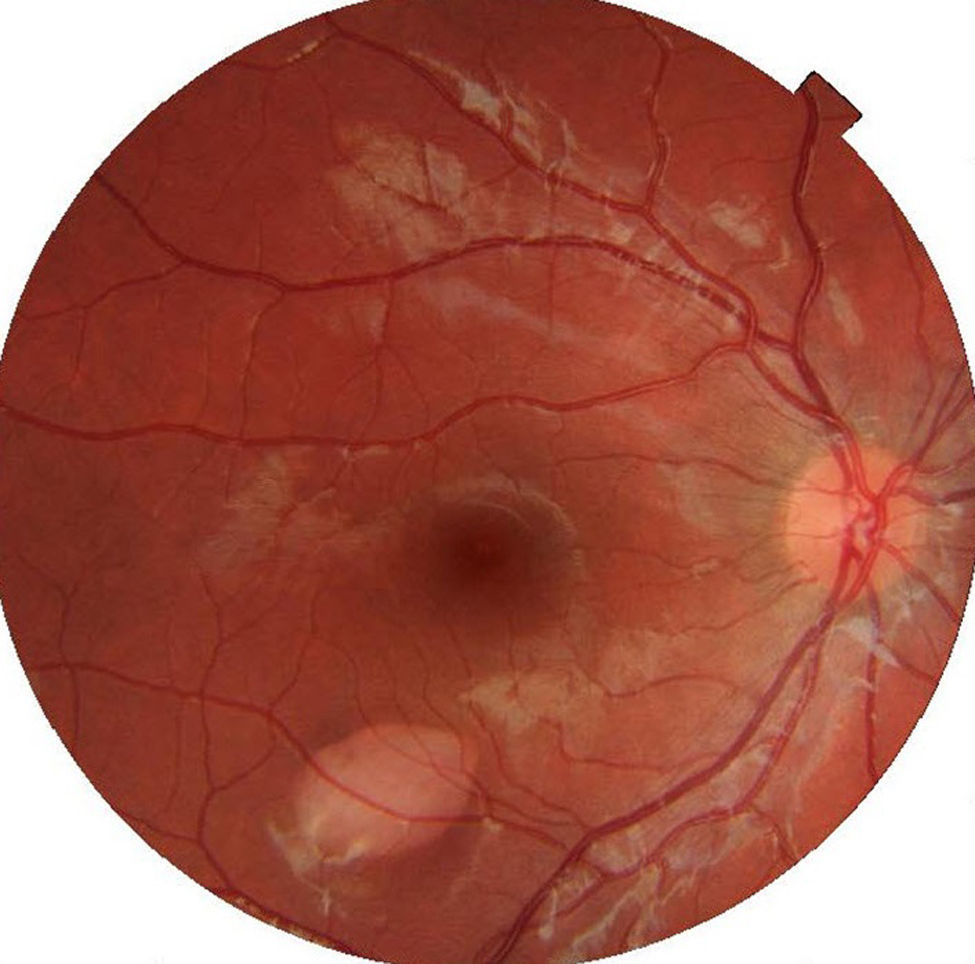
Ophthalmic examination showed the anterior segment to be normal in both eyes. The right eye presented an oval-shaped hypopigmented lesion in the lower temporal sector of the macula, pointing to the fovea, and measuring 1.7mm vertically and 2.3mm horizontally (Fig. 1).

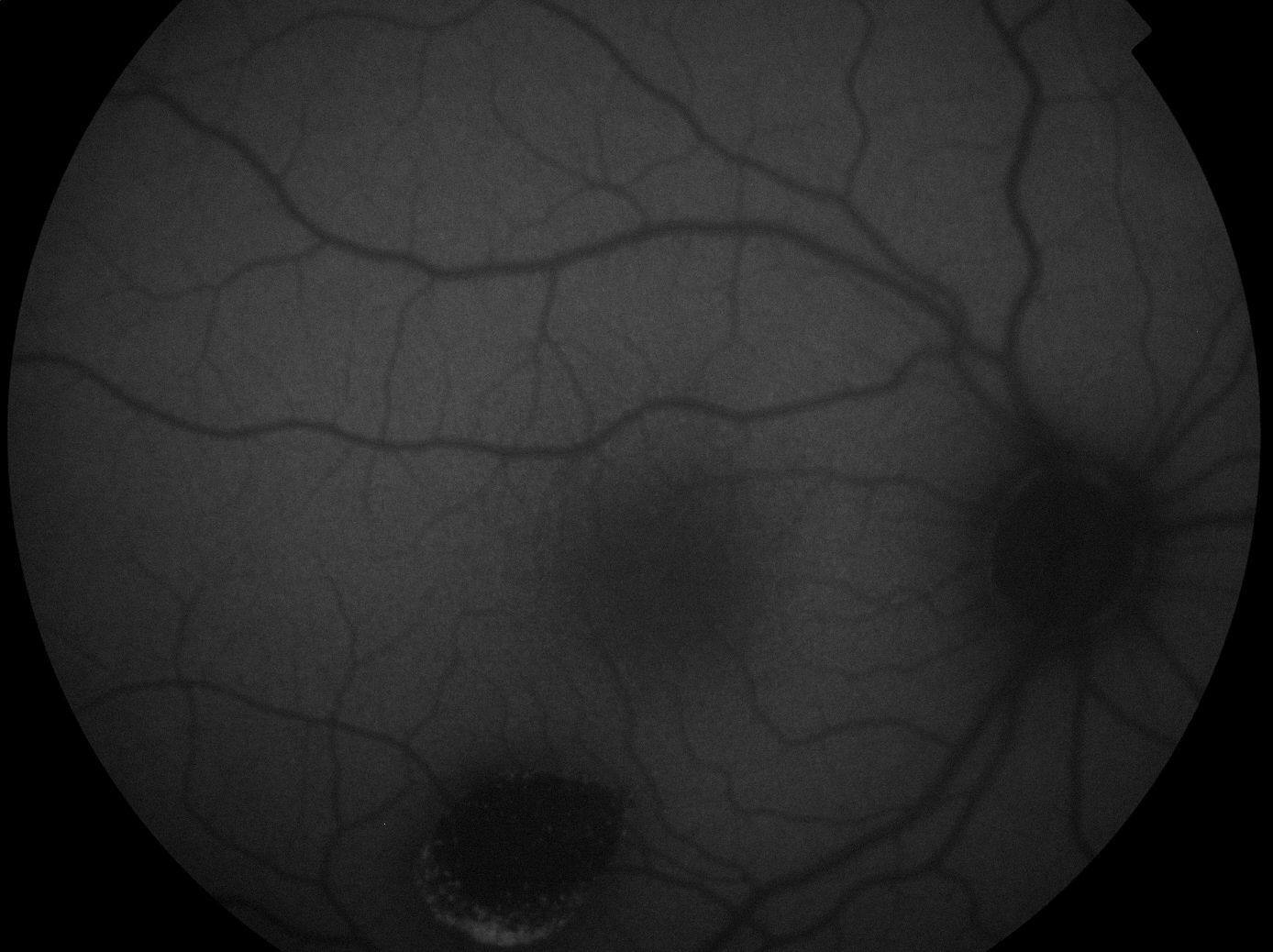
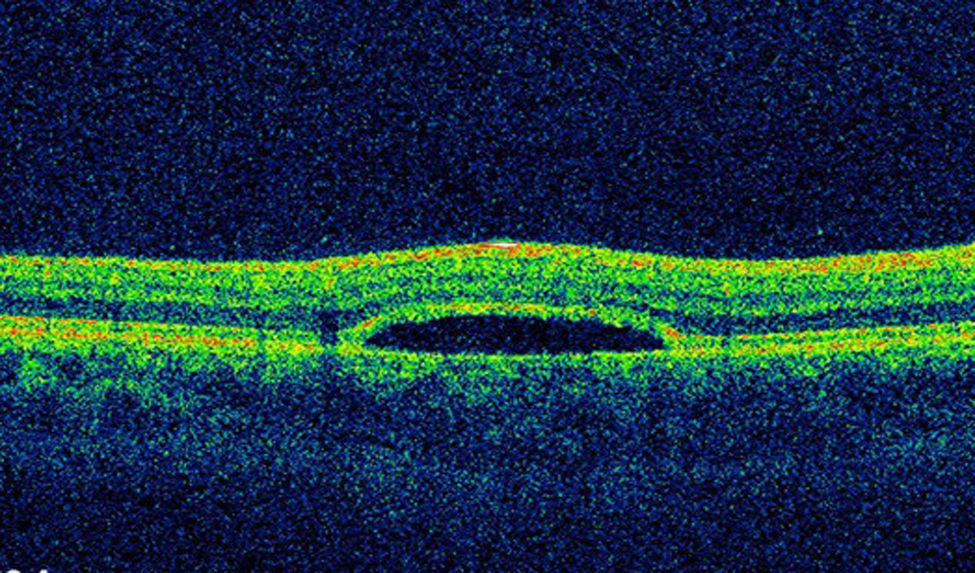
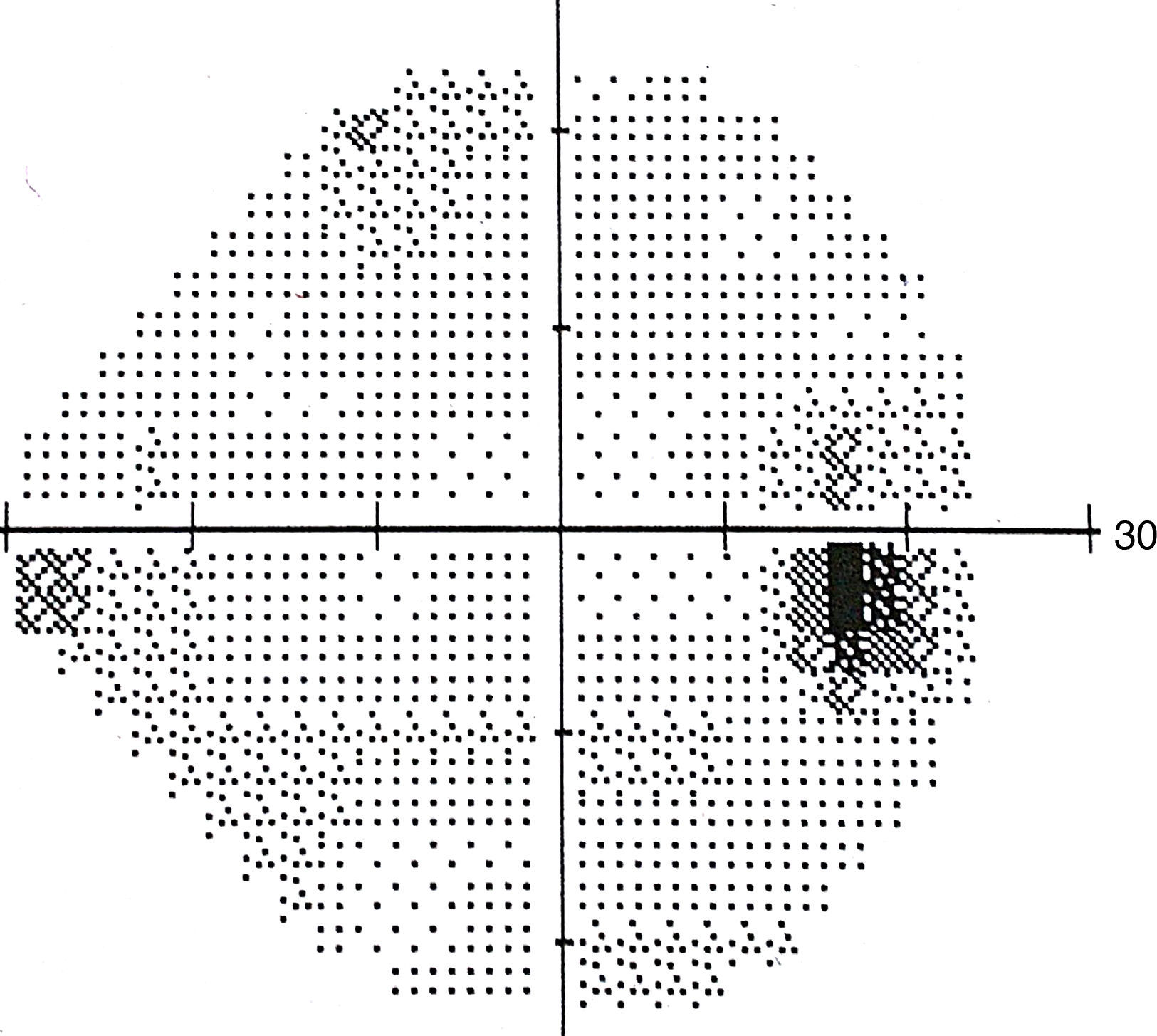
The lesion was hypo-autofluorescent with a hyper-autofluorescent lower edge (Fig. 2). Optical coherence tomography (OCT) revealed detachment of the neurosensory retina, limited to the proximity of the lesion, and reduced reflectivity of the retinal pigment epithelium (RPE). The outer layers showed no abnormalities except for the presence of scattered micro-verrucae generated by the outer segments of the photoreceptors, suggestive of chronic serous detachment of the macular neuroepithelium. Despite the lack of optimal equipment for OCT of the choroid, the reduced presence of choriocapillaris in the lesion was also observed (Fig. 3). The visual field study (Humphrey 24-2 visual field perimetry) performed two years after diagnosis revealed a relative scotoma consistent with the area of the lesion (Fig. 4). The electrooculogram and Amsler grid findings were normal.


 test result, showing a corresponding scotoma.")
After 36 months of follow up, the patient remained asymptomatic with visual acuity of 1 in both eyes, and no change according to all diagnostic techniques.
DiscussionTM is an asymptomatic lesion with a characteristic morphology that is usually detected by chance in a funduscopic examination.1 It is considered benign and there is no risk of it producing alterations in vision. In cases of decreased visual acuity there is usually another cause.4
Diagnosis of TM is basically clinical,3,11 when a fundus examination reveals a hypopigmented lesion, torpedo shaped (longer horizontally than vertically), located in the temporal area of the macula and remaining stable over time. At the nasal boundary, a tail points towards the fovea.4,9–11
Complementary tests help confirm the diagnosis.9 OCT does not produce consistent results in the area of the lesion. The RPE may appear much thinner than normal, with virtually no signal. The inner and outer retina are usually normal but may present thinning, with the loss of some outer segments of the photoreceptors.4 Subretinal fluid may be observed, possibly related to functional impairment of the RPE, with detachment of the neurosensory retina.11 In our case, there was no autofluorescence in the lesion, probably due to an absence of fluorophore compounds. This would indicate a missing or almost non-functional RPE, although Pilotto et al. described a case with normal autofluorescence.1 Fluorescein angiography revealed hyperfluorescence due to window defect, which suggests the absence of RPE.3,4 Perimetry may be normal or present a scotoma corresponding to the area of the lesión.1,4,9 In our case, the EOG, ERG, Amsler grid and colour vision test results were all normal.9
To the best of our knowledge, no histopathologic studies have been reported of TM and its aetiology is still poorly understood. However, in view of the uniformity of cases, it is assumed to be a congenital entity that appears at a very specific moment during the development of the RPE.2,3,8,9 According to Pian, it originates from a defect in the horizontal raphe during the development of the nerve fibre layer in the early postnatal period. On the other hand, Teitelbaum believes it is caused by a vascular alteration of the macular area during embryonic development, which would result in an abnormal RPE in this region.12 Given the uniform shape and location of the lesion, Shields suggested it arises from a persistent alteration during RPE development in the foetal temporal bulge.2
Differential diagnosis should be performed in conjunction with other RPE lesions such as choroidal nevi or melanomas, congenital hypertrophy of the RPE, lesions related to Gardner's syndrome or familial adenomatous polyposis síndrome.3,8–10
Pilotto et al. described the case of a 4-year-old girl with TM; autofluorescence was normal, which was ascribed to weak autofluorescence in the whole retina, given the scant presence of lipofuscin in such a young child.1 In our case, although the patient was the same age, the lesion observed presented hypo-autofluorescence, which is consistent with other published cases. Shields et al. described the case of a girl aged three years, but made no reference to autofluorescence.2 We observed no alterations in the nerve fibre layer, which contradicts Pian's hypothesis. Similarly, we observed no scarcity of RPE in the lesion, and therefore it cannot be considered a paramacular coloboma.
ConclusionGiven the benign nature of the injury, its clinical management is limited to an annual test of visual acuity, a fundus examination and an OCT to confirm the absence of changes.
Differential diagnosis should be performed in conjunction with other RPE lesions such as choroidal nevi or melanomas, congenital hypertrophy of the RPE, lesions related to Gardner's syndrome or familial adenomatous polyposis síndrome.
Ethical disclosuresProtection of human and animal subjectsThe authors declare that no experiments were performed on humans or animals for this study.
Confidentiality of dataThe authors declare that no patient data appear in this article.
Right to privacy and informed consentThe authors declare that no patient data appear in this article.
Conflict of interestThe authors have no conflict of interest or financial disclosures to report.
