


Se presenta el caso de una mujer de 48 años de edad, que inicia su cuadro clínico con disminución súbita de agudeza visual acompañado de la presencia de un escotoma central en ojo derecho unilateral, sin otros síntomas acompañantes, antecedentes personales patológicos negados, 4 semanas después la paciente recupera visión llegando a un 20/20 sin la presencia del escotoma. La epiteliopatía pigmentaria placoide posterior multifocal aguda (EPPPMA) es una patología inflamatoria coriorretiniana, que se presenta en adultos jóvenes, sanos, sin predominio por algún género, produciendo alteraciones visuales agudas, con hallazgos fundoscópicos característicos de lesiones placoides blanco-amarillentas a nivel del epitelio pigmentario de la retina. El diagnóstico se basa en los datos clínicos y la evolución complementado con la fluorangiografía, la mayoría de los casos con buen pronóstico visual, con una recuperación de la agudeza visual completa dentro de las primeras 3-6 semanas.
We present the case of a 48-year-old woman, who started with sudden decrease of visual acuity accompanied by the presence of a central scotoma in the right eye unilateral; with no other symptoms or past medical history. Four weeks later, the patient recovered 20/20 visual acuity and the scotoma had disappeared. The acute posterior multifocal placoid pigmentary epitheliopathy is an inflammatory corioretinal disease, which affects young adults, predominantly healthy women, causing acute visual alteration with fundoscopic typical findings of white-yellowish placoid lesions at the retinal pigment epithelium. The diagnosis is based on the clinical evaluation and the angiography with fluorescein; in most cases, in most cases, the visual prognosis is good, with a complete recovery of visual acuity from 3 to 6 weeks.
La epiteliopatía pigmentaria placoide posterior multifocal aguda (EPPPMA) o acute posterior multifocal placoid pigment epitheliopathy (APMPPE) por sus siglas en inglés, se define como una patología inflamatoria coriorretiniana autolimitada de etiología desconocida.
Descrita por primera vez en 1968 por Gass quien describió el cuadro clínico y los hallazgos angiográficos encontrados en 3 mujeres jóvenes presentando disminución súbita de la agudeza visual en uno o ambos ojos, lesiones en placa color blanco-amarillentas en polo posterior a nivel del epitelio pigmentario de la retina, las cuales desaparecieron ocasionando una lesión pigmentaría con mejoría de la agudeza visual1.
La EPPPMA afecta principalmente pacientes jóvenes entre la 2-4a décadas de la vida, afecta a ambos géneros por igual, generalmente sanos, la enfermedad es clásicamente bilateral y solo algunos casos aislados se han descrito de forma unilateral2–4.
Estos pacientes generalmente inician con baja visual asimétrica, acompañada de un escotoma central, también se pueden presentar fotopsias, visión borrosa y miodesopsias siendo un cuadro autolimitado con una mejoría de la agudeza visual total en 3-6 semanas.
Durante la fundoscopía es característico encontrar lesiones placoides blanco-amarillentas a nivel del epitelio pigmentario de la retina con predominio en polo posterior, en la fluorangiografía el patrón característico consiste en hipofluorescencia en etapas iniciales con hiperfluorescencia en fases tardías5.
La EPPPMA en general no requiere tratamiento ya que la recuperación es espontánea en la mayoría de los casos con recuperación de la agudeza visual. Algunos autores sugieren el uso de esteroides en casos asociados a vasculitis, papilitis o con involucro macular6.
Aunque a nivel ocular la enfermedad es en términos generales benigna, es importante mencionar que puede estar asociada a alteraciones neurológicas siendo la más frecuente cefalea, otros síntomas descritos son parestesias, psicosis, vértigo y algunas más severas como evento vascular cerebral secundario a vasculitis7.
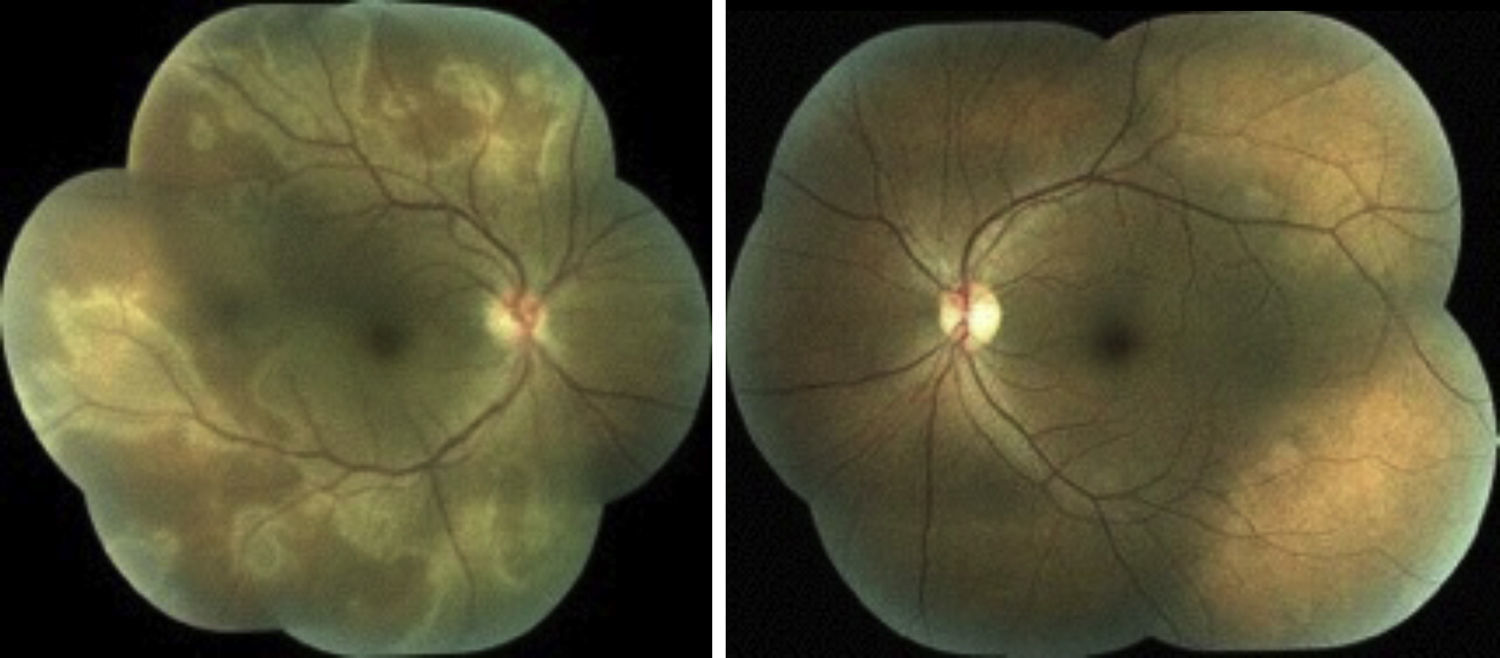
Presentación del casoPaciente del sexo femenino, de 48 años de edad, maestra, que acude a consulta por presentar disminución de la agudeza visual en ojo derecho de forma súbita de 3 días de evolución sin ningún síntoma acompañante. Antecedentes personales patológicos negados. A la exploración oftalmológica presenta una agudeza visual del ojo derecho (OD) 20/60 que no mejora con estenopeico, ojo izquierdo (OI) 20/20, se realiza refracción reportando OD +1.00 con 0.50100° y (OI) +0.62 con 0.3795°, capacidad visual del OD 20/60 y del OI 20/20; se realiza amsler encontrando un escotoma central en ojo derecho. Presión intraocular 16mmhg en ambos ojos. En segmento anterior se observa cornea transparente, cámara anterior formada, pupila redonda, refléctica, cristalino transparente en ambos ojos, tyndall negativo. En segmento posterior en ojo derecho encontramos excavación de 0.3, emergencia central de vasos, la presencia de lesiones placoides blanco-amarillentas que involucran polo posterior y se extienden fuera de las arcadas, retina aplicada, papila de bordes sutilmente difusos, vítreo transparente. Ojo izquierdo de características normales (fig. 1).

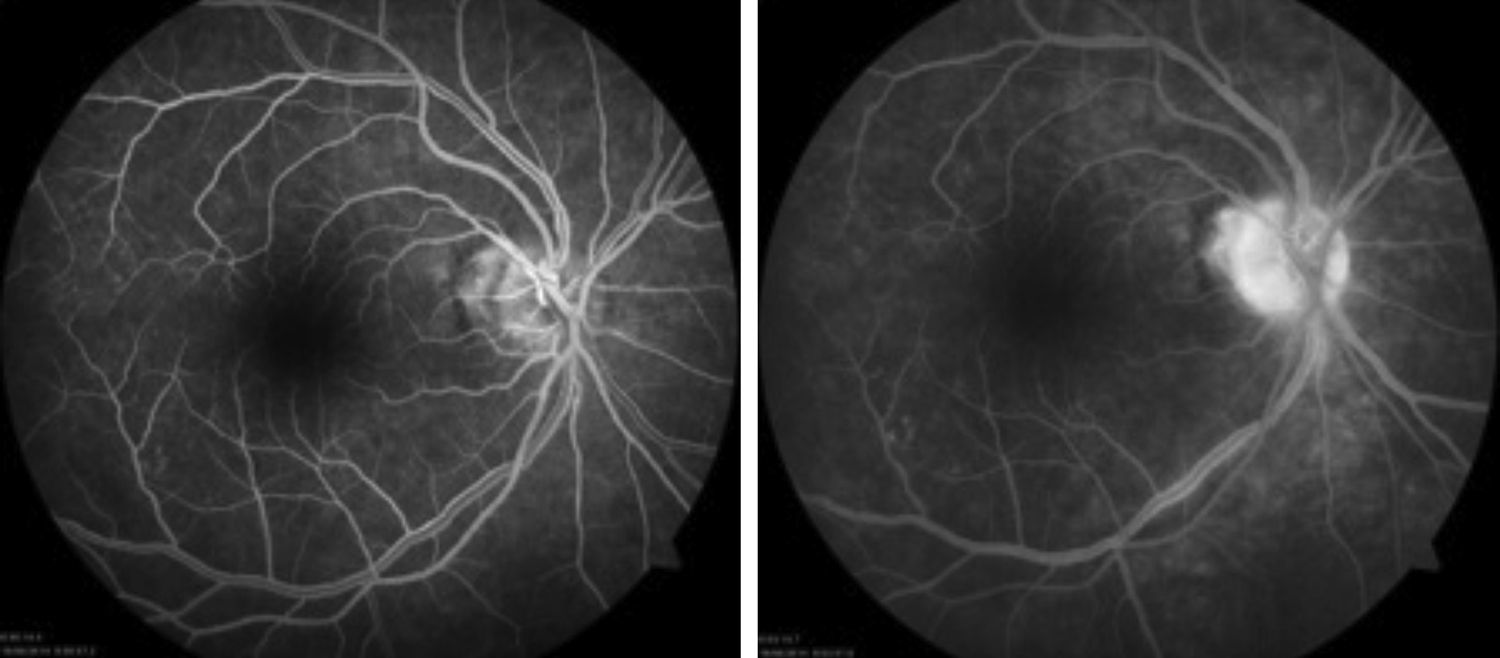
La angiografía con fluoresceína se realizó con una cámara digital de alta resolución del fondo de ojo ZEISS, VISUCAM, Carl Zeiss Meditec A6, 07740, Jena, Alemania. Se observó ojo derecho en la fase arterio-venosa media fluorescencia homogénea en los 4 cuadrantes; en la fase tardía se aprecia una hiperfluorescencia papilar (fig. 2).

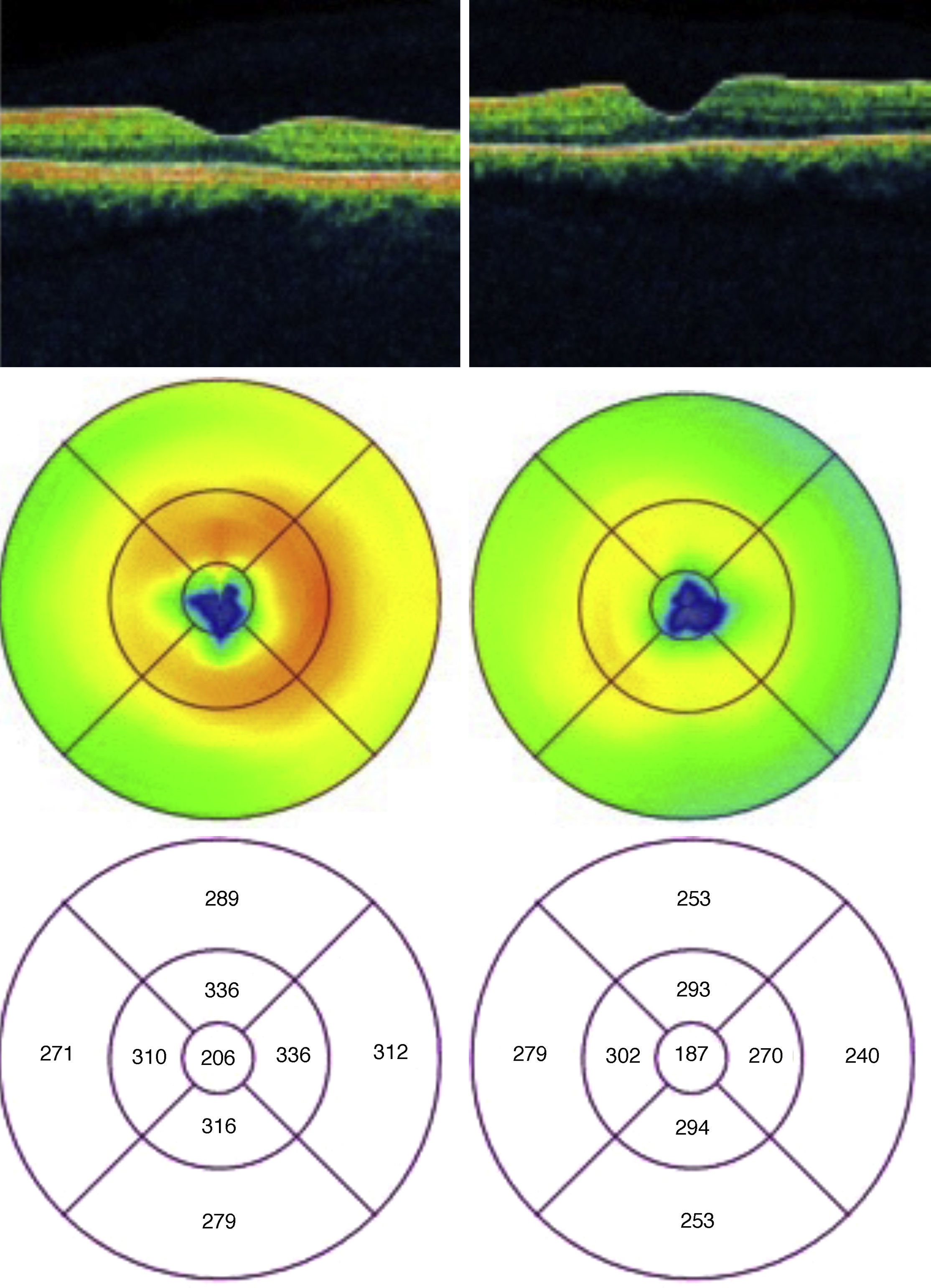
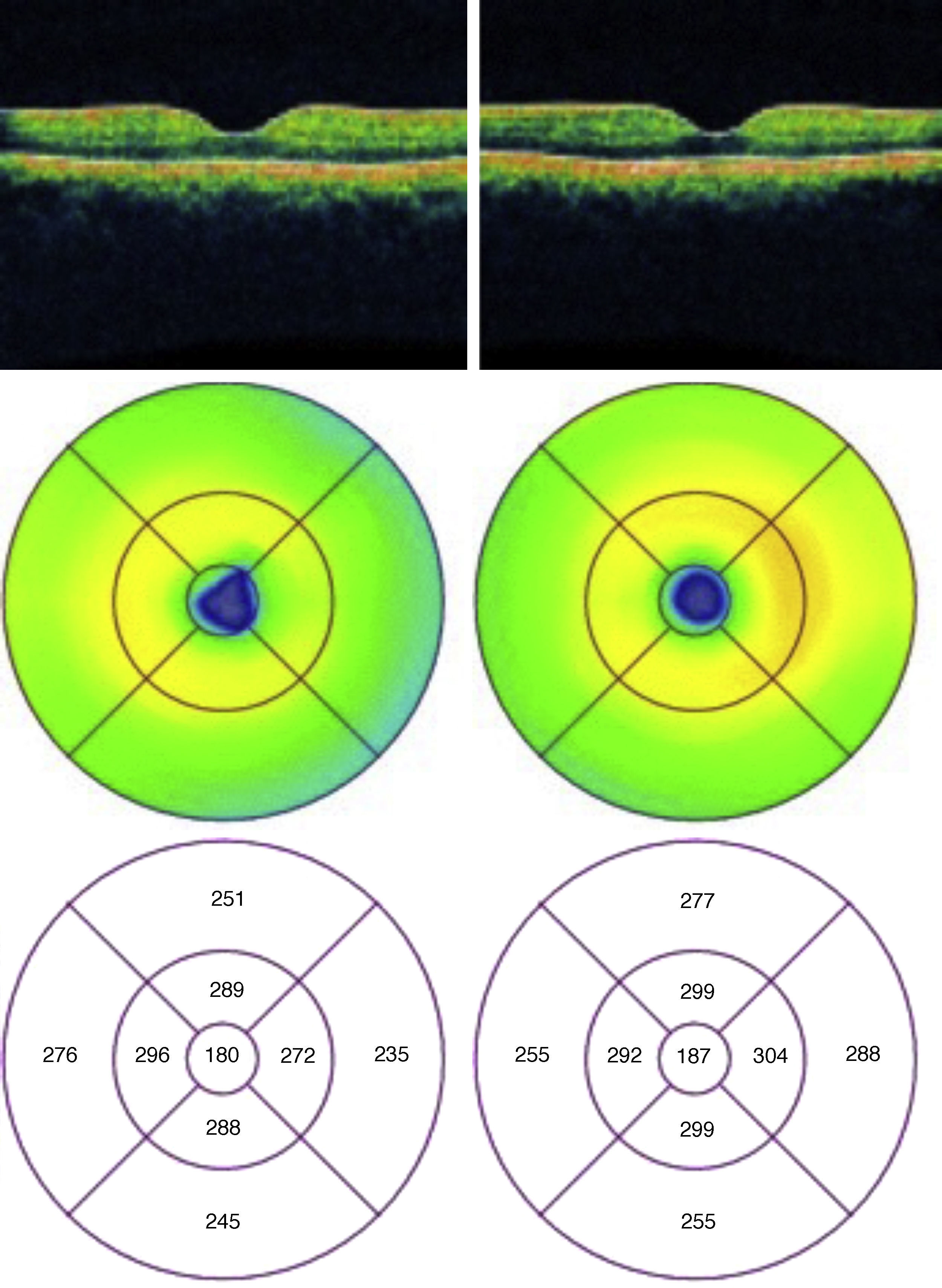
Se solicitó tomografía de coherencia óptica (TCO) ambos ojos que se realizó con el equipo de TCO tipo time-domain (TD-TCO) Stratus; Carl Zeiss Meditec, Inc. En el mapa macular, en el círculo concéntrico de los 3mm se aprecia un aumento del grosor retiniano en ojo derecho, ojo izquierdo de características normales (fig. 3).

Se solicitan marcadores inflamatorios básicos velocidad de sedimentación globular (10mm/h), proteína C reactiva (0.1mg/dL), factor reumatoide (20u/ml) se reportaron dentro de parámetros normales, anticuerpos antinucleares 1:40 negativo, estudios serológicos para VIH y VDRL resultaron negativos. Debido al hallazgo de papilitis se solicitó una resonancia magnética de cráneo en la cual no se observaron zonas hipointensas sugestivas de vasculitis.
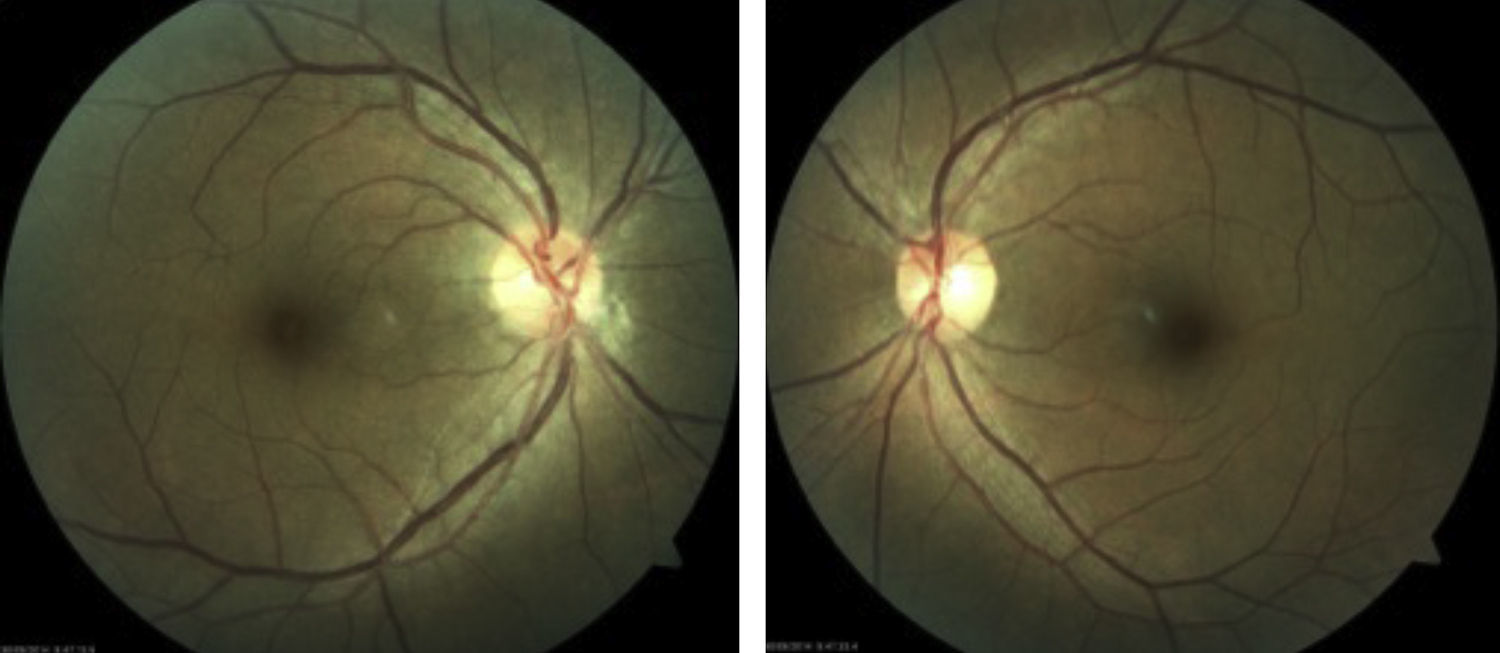
Una vez descartada alguna patología del sistema nervioso central asociada con el hallazgo de papilitis se decide iniciar con esteroides vía oral. Cuatro semanas después de haber iniciado el cuadro clínico, la agudeza visual en ojo derecho se recuperó, alcanzando una agudeza visual de 20/20, desapareciendo el escotoma central. A la exploración de fondo de ojo no se aprecian las lesiones placoides blanco-amarillentas con normalización de bordes papilares en ojo derecho. Ojo izquierdo de características normales (fig. 4).

Se decide realizar una nuevamente tomografía de coherencia óptica un mes después de ambos ojos, en donde se aprecia una disminución en el volumen macular en el círculo de los 3mm en el ojo derecho (fig. 5).

Se realiza campimetría posterior al tratamiento con un equipo Humphrey Visual Analyzer, Carl Zeiss Meditec Inc.; Oberkochen, Alemania. Prueba SITA-Standard 30-2 confiable, reportándose dentro de límites normales en ambos ojos, no se observa escotoma en ojo derecho.
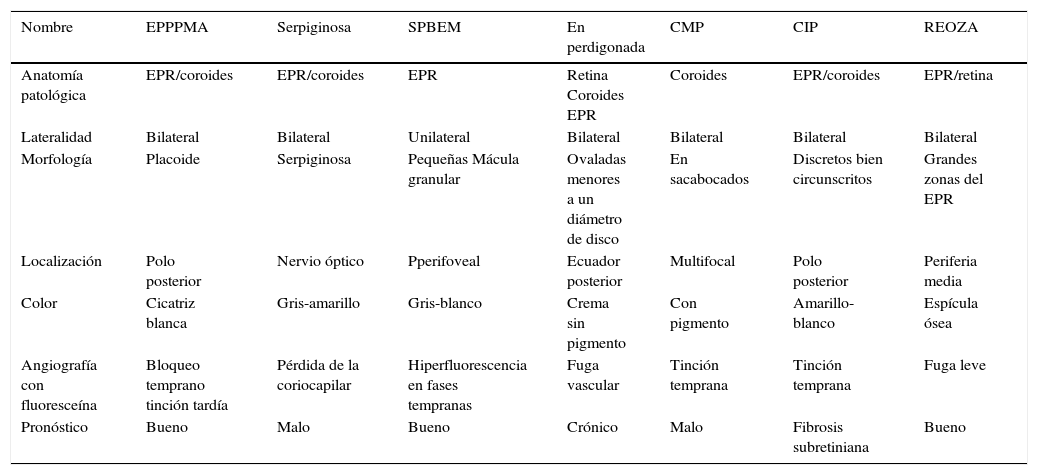
En cuanto al diagnóstico diferencial existen varias patologías que presentan un cuadro similar los cuales se engloban en el término de síndromes de puntos blancos que engloba a la EPPPMA, síndrome de puntos blancos evanescentes múltiples (SPBEM), coroiditis serpiginosa, retinocoroidopatía en perdigonada, síndrome de coroiditis multifocal y panuveítis (CMP), coroidopatía interna punteada (CIP) y retinopatía aguda zonal oculta externa (REOZA). En la (tabla 1) se mencionan las características principales de cada uno de estos síndromes.
Síndromes de puntos blancos
| Nombre | EPPPMA | Serpiginosa | SPBEM | En perdigonada | CMP | CIP | REOZA |
|---|---|---|---|---|---|---|---|
| Anatomía patológica | EPR/coroides | EPR/coroides | EPR | Retina Coroides EPR | Coroides | EPR/coroides | EPR/retina |
| Lateralidad | Bilateral | Bilateral | Unilateral | Bilateral | Bilateral | Bilateral | Bilateral |
| Morfología | Placoide | Serpiginosa | Pequeñas Mácula granular | Ovaladas menores a un diámetro de disco | En sacabocados | Discretos bien circunscritos | Grandes zonas del EPR |
| Localización | Polo posterior | Nervio óptico | Pperifoveal | Ecuador posterior | Multifocal | Polo posterior | Periferia media |
| Color | Cicatriz blanca | Gris-amarillo | Gris-blanco | Crema sin pigmento | Con pigmento | Amarillo-blanco | Espícula ósea |
| Angiografía con fluoresceína | Bloqueo temprano tinción tardía | Pérdida de la coriocapilar | Hiperfluorescencia en fases tempranas | Fuga vascular | Tinción temprana | Tinción temprana | Fuga leve |
| Pronóstico | Bueno | Malo | Bueno | Crónico | Malo | Fibrosis subretiniana | Bueno |
CIP: coroidopatía interna punteada; CMP: síndrome de coroiditis multifocal y panuveítis; EPR: epitelio pigmentario de la retina; EPPPMA: epiteliopatía pigmentaria placoide posterior multifocal aguda; REOZA: retinopatía aguda zonal oculta externa; SPBEM: síndrome de puntos blancos evanescentes múltiples.
Si hablamos de la EPPPMA se cree que existen más de 200 casos reportados, esta patología presenta predominio por adultos jóvenes, sin predominio establecido por algún genero2. La EPPPMA característicamente se presenta con disminución de la agudeza visual, y esta puede acompañarse por escotomas o metamorfopsias, en 75% de los casos es bilateral aunque esta descrito que puede comenzar de forma unilateral y en el transcurso de meses o años afectar el ojo contralateral2. En la fundoscopía es característico encontrar lesiones placoides blanco-amarillentas, con predominio en polo posterior2. En algunos casos también está reportada la presencia de papilitis; Kirkham et al. sugieren que la presencia de papilitis en EPPPMA probablemente refleje involucro del flujo coroideo hacia el nervio óptico, lo cual sugiere un peor pronóstico visual y en estos casos puede estar indicado el uso de esteroides6. En la fluorangiografía se observan lesiones hipofluorescentes en etapas tempranas con hiperflourescencia en etapas tardías2,8. Lo que sugiere una inadecuada perfusión coroidea. La etiología de la EPPPMA es aún incierta, existe la posibilidad de que la etiología sea infecciosa por la frecuencia con que se asocia a un cuadro gripal9–11. Se considera que la enfermedad tiene una etiología inflamatoria y que el mecanismo desencadenante de la misma es la activación de los leucocitos por un proceso infeccioso. Se han reportado múltiples asociaciones como uveítis, desprendimiento de retina seroso, vasculitis9, inmunizaciones (influenza, varicela, hepatitis b), granulomatosis de Wegener2,4 y algunos subtipos de HLA-A3, A2, B7 y C74,12.
En relación con el tratamiento se trata de una patología autolimitada la mayoría de las ocasiones con buen pronóstico visual por lo que no se sugiere tratamiento, aunque algunos autores reportan que está justificado el uso de esteroides en casos con involucro del sistema nervioso central, macular o papilitis4,13.
En nuestra paciente se presentó un cuadro clásico de EPPPMA, con disminución súbita de la agudeza visual acompañada de escotoma central, sin ningún otro síntoma acompañante, en este caso unilateral. En la fundoscopía se encontraron lesiones placoides blanco-amarillentas características en este caso asociadas a papilitis, con mejoría de la agudeza visual a las 4 semanas y desaparición de las lesiones clínicas sin dejar secuelas.
ConclusionesLa EPPPMA es una entidad poco frecuente que en muchas ocasiones no es diagnosticada debido a su curso autolimitado, generalmente es bilateral y la mayoría de los casos con buen pronóstico visual, en este caso concluimos en este diagnóstico ya que la paciente presentó un cuadro característico de esta patología comenzando con baja visual, que si bien no es lo clásico que sea unilateral, existen casos reportados en la literatura, las lesiones placoides blanco-amarillentas en polo posterior de causa no determinada que se autolimitaron de forma espontánea sin presentar secuelas con una recuperación completa de la agudeza visual.
Responsabilidades éticasProtección de personas y animalesLos autores declaran que para esta investigación no se han realizado experimentos en seres humanos ni en animales.
Confidencialidad de los datosLos autores declaran que han seguido los protocolos de su centro de trabajo sobre la publicación de datos de pacientes.
Derecho a la privacidad y consentimiento informadoLos autores han obtenido el consentimiento informado de los pacientes y/o sujetos referidos en el artículo. Este documento obra en poder del autor de correspondencia.
FinanciamientoLos autores no recibieron patrocinio para llevar a cabo este artículo.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.
