


La enfermedad de Devic es una rara patología desmielinizante que afecta al nervio óptico y que se acompaña de sintomatología medular. A continuación se presenta el éxito terapéutico de la plasmaféresis en este caso, como el primero en Ecuador; asimismo caracterizamos el cuadro y analizamos el beneficio del tratamiento en la historia natural de la enfermedad. Se trata de un paciente masculino, adulto, con antecedente de amaurosis súbita, y que comienza con una mielopatía aguda. En ausencia de trauma, se investigaron otras causas y se concluyó con el diagnóstico clínico e imagenológico de neuromielitis óptica. Consecuentemente se consiguió la mejoría en síntomas medulares de sensibilidad y función motora a corto plazo.
Devic's disease is a rare demyelinating pathology that affects the optic nerve and spinal cord; following we describe the successful treatment with plasmapheresis of this condition as the first in Ecuador; likewise characterize and analyze the benefits of treatment on the natural history of the disease. The case is a male patient, adult, with a history of sudden amaurosis, which debuts with acute level spinal alteration; in the absence of trauma, other causes are investigated and concluded with the clinical and imaging diagnosis of neuromielytis optica. The results are the success in medullar symptoms in sensitivity and motor functions.
La neuromielitis óptica (NMO) es una entidad que combina neuritis del nervio óptico y mielitis transversal; es una patología de sustancia blanca y su etiología es autoinmune. Hace algunos años fue considerada como una forma de esclerosis múltiple (EM), sin embargo posee una clínica, serología e imagenología particular. Eugene Devic (Lyon, 1858-París, 1930) caracterizó la enfermedad en 18941.
El caso fue investigado en el servicio de clínica del Hospital Vicente Corral Moscoso durante un periodo de 8 meses. Se trata de un paciente masculino, procedente del cantón Pucará, Azuay, Ecuador, sin factores de riesgo, ni antecedentes familiares ni personales, cuyo cuadro comenzó con amaurosis súbita y después de un periodo de varios años presenta sintomatología medular. Al inició se trató con corticoterapia de rescate e inmunomodulación, después del diagnóstico etiológico como una recaída, decidiéndose la plasmaféresis y evaluando su resultado.
Presentación del caso clínicoPaciente de 45 años, quien se dedicaba a la vigilancia de seguridad hace 7 meses, refiere hace 9 años sin causa aparente amaurosis súbita del ojo derecho, sin síntomas acompañantes; por este motivo fue atendido en el servicio de oftalmología, diagnosticándole infección ocular. Hace 7 meses presenta disminución de la agudeza visual en el ojo izquierdo que empieza en la mañana y culmina con amaurosis izquierda ese mismo día, considerándola como una nueva infección ocular; después de 3 semanas recupera breve agudeza visual de lado izquierdo observando solo sombras y colores básicos. Aproximadamente 4 días antes del ingreso presenta parestesias intensas que descienden desde nivel intercostal 7.o hacia todo el miembro inferior izquierdo; al cuadro se le suma paresia progresiva al lado izquierdo y parestesias leves que ascienden desde pie derecho hasta nivel intercostal 7.o del lado derecho; también en ese momento presenta hipoestesia desde rodilla hacia sus dedos solo de la parte anterior y dorso del pie del mismo lado. La sintomatología se exacerba con impotencia funcional de ambos miembros inferiores, impidiendo la deambulación.
Las alteraciones en sus signos vitales en el ingreso fueron: presión arterial de 130/60mm/Hg y taquicardia de 118lpm.
Se analiza el examen neurológico y oftalmológico, en los que se evidencia lo siguiente:
- •
Funciones mentales superiores: vigil, orientado en tiempo, espacio y persona. Memoria retrógrada, anterógrada, atención, cálculo, lenguaje, praxias y gnosias sin alteración.
- •
Nervios craneales:
- ∘
II (nervio óptico):
- 1.
Agudeza visual cercana: evaluación por cartelón de Rosenbaum, no distingue ninguna letra con ninguno de los ojos.
- 2.
Agudeza visual lejana: tabla de Snellen: ceguera en ojo izquierdo: 20/200; ceguera total en ojo derecho. Cuenta dedos: distingue con ojo izquierdo. Movimiento de manos: sí distingue con ojo izquierdo. Percepción y discriminación de luz: no discrimina ni percibe luz con ojo derecho y sí con izquierdo, pero el paciente refiere que empeora la agudeza de ojo izquierdo ante el calor (fenómeno de Uhthof).
- 3.
Visión colores: ninguna percepción de colores con ojo derecho y con ojo izquierdo percepción de colores primarios. (evaluación por tabla de Ishihara).
- 4.
Campimetría por confrontación: preserva visión periférica y central en ojo izquierdo. En derecho, no preservada.
- 5.
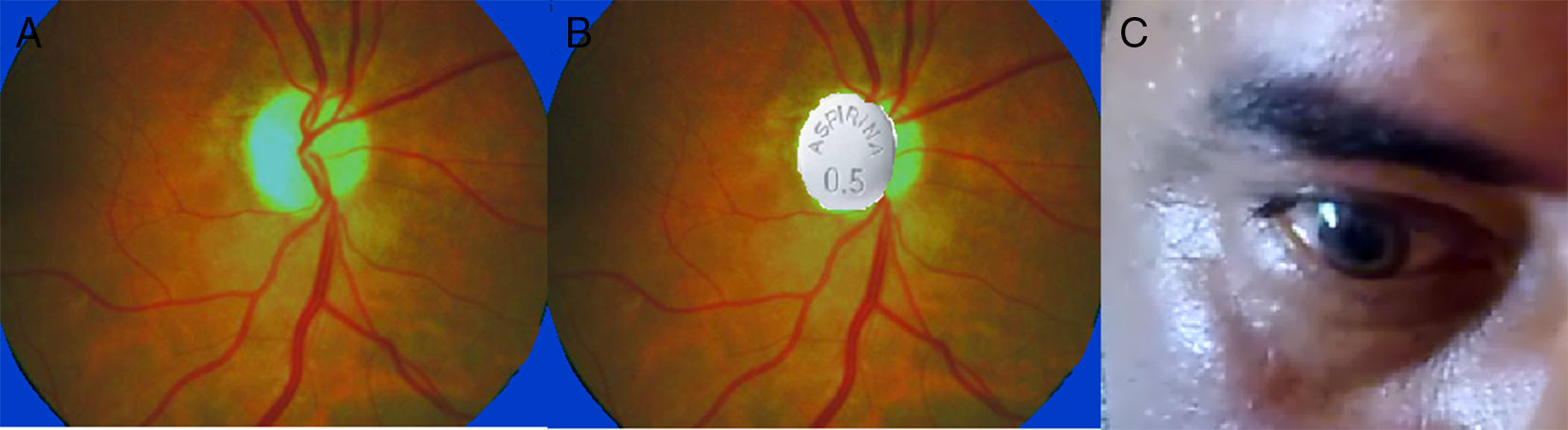
Fondo de ojo: papila pálida y atrófica bilateral (en «aspirina») (fig. 1).
- 1.
- ∘
III, IV, VI: movilidad extraocular conservada; movilidad consensuada de ojo izquierdo con respuesta del derecho y del derecho sin respuesta del izquierdo. Reflejo fotomotor directo: pupilas midriáticas e hiporreactivas bilaterales. Reflejo de acomodación consensuado sin respuesta en ojo derecho (fig. 1).
- ∘
V, VII, VIII, IX, X, XI, XII: normales sin alteración.
- •
Sistema motor: fuerza muscular: extremidades superiores: 5/5; extremidades inferiores: –derecha: 4/5, –izquierda: 2/5. Tono muscular normal en las 4 extremidades.
- •
Reflejos de estiramiento muscular: hiperreflexia rotuliana bilateral (L2-L4, inclusive al tacto), hiporreflexia aquileana bilateral (S1). Presencia de Babinski y sucedáneos bilaterales. Presencia de clonus del tobillo izquierdo agotable (6-8 oscilaciones).
- •
Sensibilidad superficial y profunda:
- ∘
- ∘
Dolor y temperatura (haces espinotalámicos): analgesia desde la rodilla derecha hacia abajo; anterior hasta dorso de los dedos del pie derecho.
- ∘
Posición y vibración (columnas posteriores): apalestesia, abarestesia desde la rodilla derecha hacia abajo; anterior hasta dorso de los dedos del pie derecho. (Compromiso de niveles: L4 y L5). Asomatognosia de miembro inferior izquierdo.
- ∘
Tacto fino (2 anteriores): disestesia desde nivel intercostal 7.o izquierdo desciende hasta pie izquierdo.

El resto del examen neurológico, regional y de sistemas fue normal.
Exámenes diagnósticos complementariosHemograma, química sanguínea, coagulación, serología e inmunología, examen elemental y macroscópico de orina, serología para el virus de la inmunodeficiencia humana, hepatitis B y C: Normales sin alteración. Examen de líquido cefalorraquídeo: Pleocitosis (neutrófilos: 10%), linfocitosis (90%) hipoproteinorraquia (0.2mg/dl), glucorraquia normal (52mg/dl). Tomografía axial computarizada de cráneo: Sin alteración.
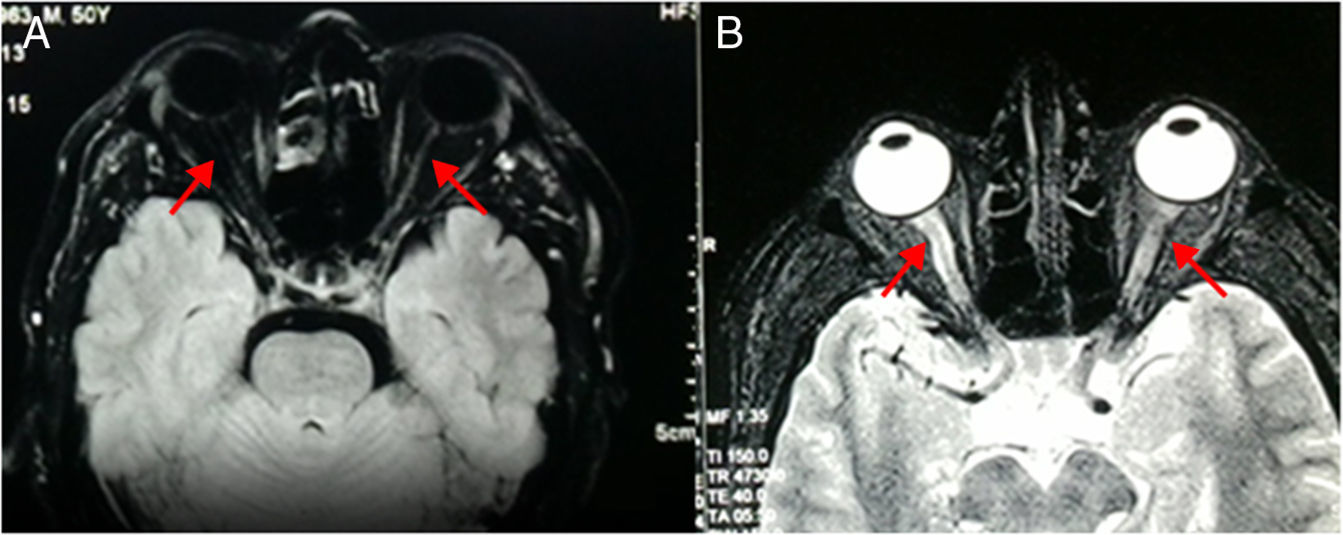
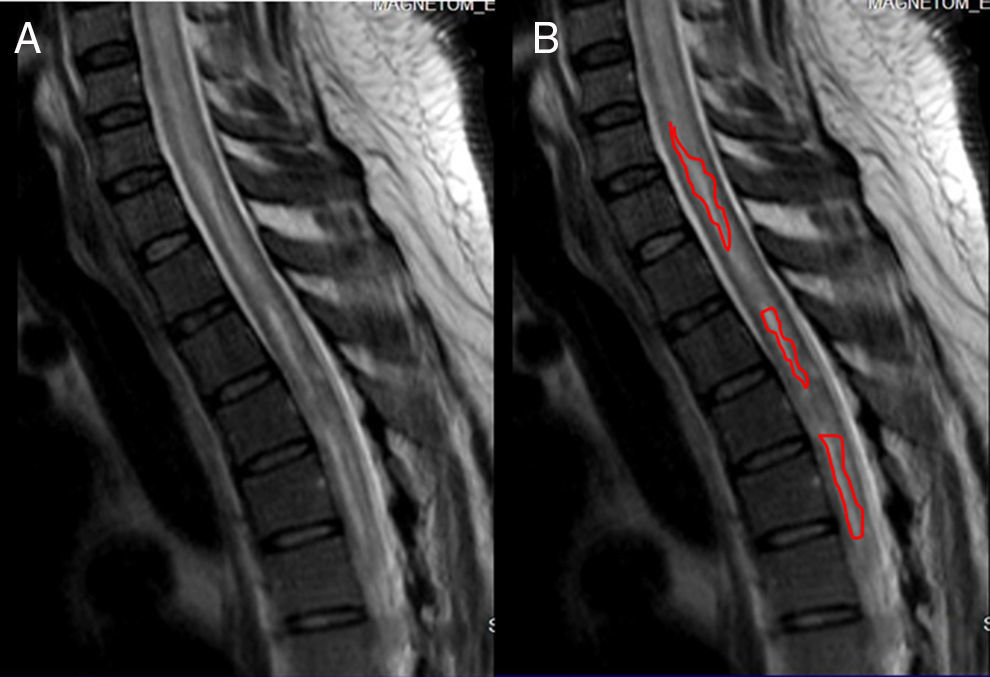
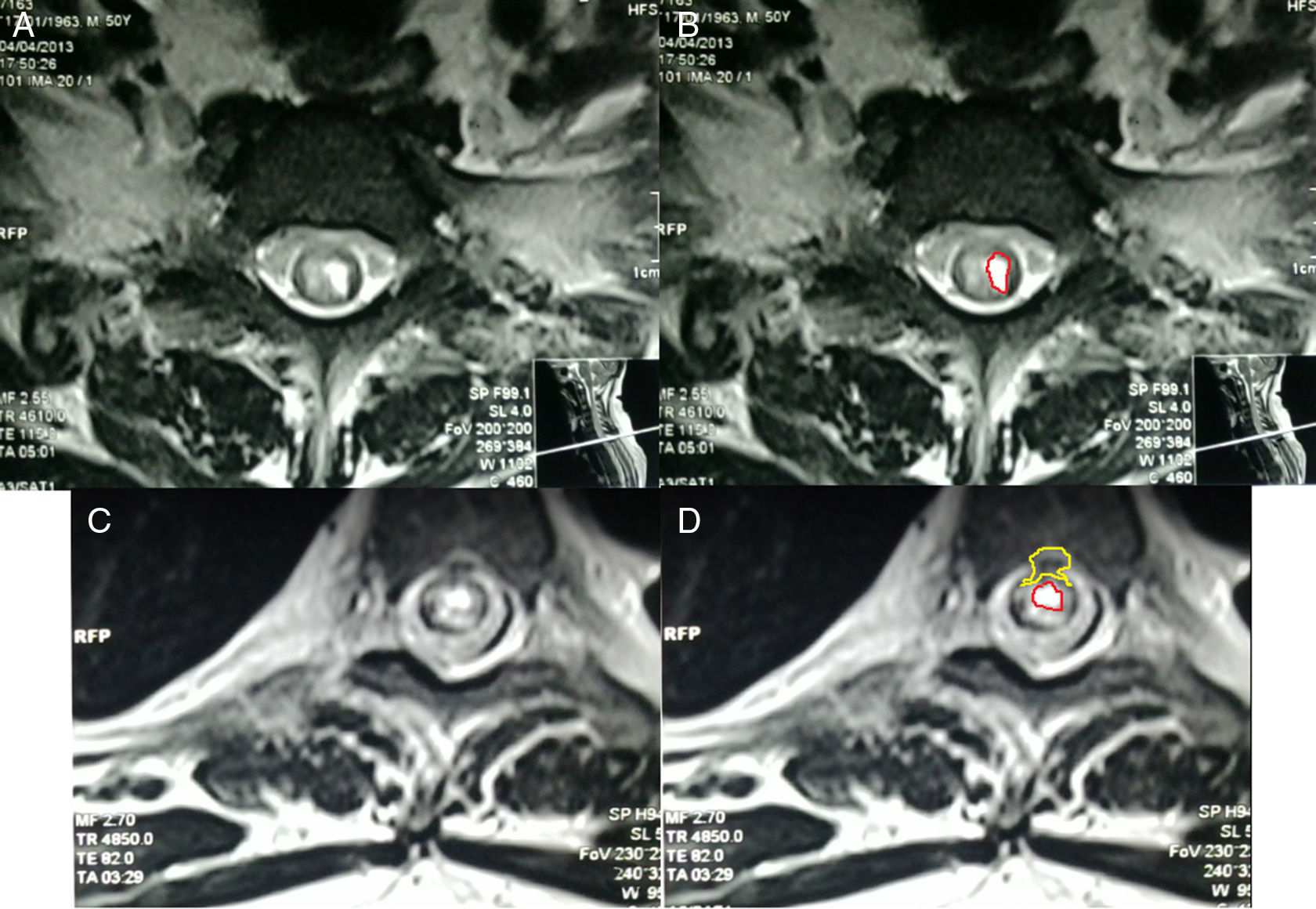
Resonancia magnética de cráneo y médula espinal: Atrofia nervio óptico derecho en su porción intraorbitaria; neuritis de nervio óptico izquierdo en la misma localización. En médula espinal se observa hiperintensidad de la señal en T2 en el interior de la médula cervical y dorsal; desde C3 hasta T7 demuestra degeneración medular extensa. Con la administración de gadolinio en T1 se observó una captación heterogénea del mismo en el interior de la médula. A nivel lumbar se ve un discreto abombamiento del disco L4-L5, secundario a inflamación, que comprime la porción anterior del saco dural (figs. 2–4).

Comparación de resonancias magnéticas de cráneo T1 y T2. Fig. 2-A: En T1 los nervios ópticos son brillantes; en el caso se ve en el lado derecho oscurecido y el izquierdo es muy delgado en «hilacho». Fig. 2-B: Resonancia realizada 7 meses antes del ingreso. En T2 los nervios ópticos son hipointensos; en el caso se ve en el lado derecho con periferia hipercaptante de señal y el izquierdo tiene trayecto interrumpido. Fuente: RM de Instituto de Diagnóstico por imagen; elaborado por los autores.


-A y B. Resonancia magnética corte transversal de médula espinal cervical T1. Se observan áreas hipercaptantes de gadolinio de médula espinal hipointensa en T1. Fig. 4-C y D. RM, corte transversal de porción lumbar donde se observa protrusión saco dural y patrón de captación de gadolinio. Fuente: RM de Instituto de Diagnóstico por imagen; elaborado por: Autores.
Potenciales visuales evocados: Sin respuesta en ojo derecho y en ojo izquierdo aumento de latencia de P100, diferencia interocular de gran variabilidad. Se demuestra alteración en la conducción de nervio óptico hacia corteza occipital.
En los diagnósticos diferenciales, se descartaron las siguientes posibilidades más frecuentes:
- •
Enfermedades desmielinizantes difusas: EM.
- •
Enfermedades con neuritis y atrofia óptica: enfermedad de Laeber, diabetes, intoxicación por alcohol metílico, arteritis de la temporal de células gigantes.
- •
Leucoencefalomielopatías: neoplásicas (linfomas de células T), hemorrágicas, infecciosas, inflamatorias, de errores innatos del metabolismo.
- •
Afectaciones mecánicas en médula y procesos traumáticos.
- •
Enfermedades con síndromes medulares secundarios: lupus eritematoso sistémico, virus de la inmunodeficiencia humana, síndromes para neoplásicos.
- •
Intoxicaciones por metales pesados.
Los criterios para identificar NMO, que precisaron el diagnóstico1 fueron:
- •
Síntomas medulares y ópticos (mielitis transversal y neuritis óptica): Cumple.
- •
Hallazgos en resonancia magnética: lesiones que no cumplan los criterios de EM, y evidencia lesión longitudinal en T2, con extensión de 3 o más segmentos vertebrales: Cumple.
- •
Seropositividad para anticuerpo patógeno específico test NMO-IgG: antiacuaporina-4 (AQP4): Cumple (tabla 1).
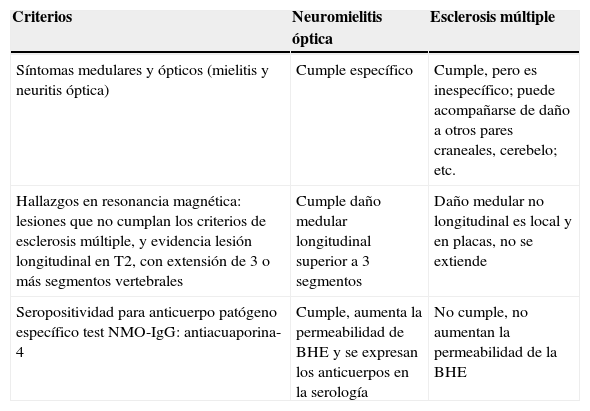
Tabla 1.Criterios diagnósticos de NMO; comparación NMO vs. EM
Criterios Neuromielitis óptica Esclerosis múltiple Síntomas medulares y ópticos (mielitis y neuritis óptica) Cumple específico Cumple, pero es inespecífico; puede acompañarse de daño a otros pares craneales, cerebelo; etc. Hallazgos en resonancia magnética: lesiones que no cumplan los criterios de esclerosis múltiple, y evidencia lesión longitudinal en T2, con extensión de 3 o más segmentos vertebrales Cumple daño medular longitudinal superior a 3 segmentos Daño medular no longitudinal es local y en placas, no se extiende Seropositividad para anticuerpo patógeno específico test NMO-IgG: antiacuaporina-4 Cumple, aumenta la permeabilidad de BHE y se expresan los anticuerpos en la serología No cumple, no aumentan la permeabilidad de la BHE Fuente: Diagnóstico de Sociedad de Ciencias Neurológicas Europeas NMO; elaborado por los autores de este trabajo.
En el ingreso se procedió a hidroterapia con solución salina, y corticoterapia con metilprednisolona, vía venosa a bolos, 1g/24h, durante 5 días. Después de realizar estudios se añadió: prednisona 50mg vía oral/24h durante un mes; azatioprina 50mg, vía oral/24h durante un mes; y ante la consideración de recaída, se decide plasmaféresis en 3 sesiones.
DiscusiónLa NMO es una enfermedad desmielinizante difusa; la Clasificación Internacional de Enfermedades (CIE-10) la describe con el código «G36», posee características, clínica, imágenes, serología e inmunopatología propias; el principal problema son los episodios de recaídas, por lo que este artículo defiende el beneficio de la plasmaféresis en este tipo de casos.
EtiologíaEs de carácter autoinmune causada por la unión de anticuerpo-AQP4 encontrada en el 70% de los casos de NMO; estos son autoanticuerpos (IgG-NMO) presentes en suero sanguíneo2,3.
FisiopatologíaEl blanco de los anticuerpos es el canal proteínico AQP4 que se encuentra en: la barrera hematoencefálica (BHE), pía, subpía, espacios de Virchow-Robin y microvasculatura de la sustancia blanca del cerebelo, mesencéfalo y médula espinal4, el canal se halla en endotelio del espacio perivascular en donde acontece la fijación del complemento y la cascada inflamatoria con macrófagos, eosinófilos y linfocitos aumentando la permeabilidad de BHE y así se expresan los anticuerpos en suero. La sustancia gris se ve alterada secundariamente por el edema y la inflamación de sustancia blanca5.
Anatomía patológicaEn la fase aguda ocurre inflamación difusa y ablandamiento que se extiende a través de múltiples segmentos medulares. El examen histopatológico revela la necrosis tanto de la materia gris y como de la blanca, con la infiltración de macrófagos asociada a la mielina y la pérdida axonal, infiltración inflamatoria perivascular variable. Tardíamente la atrofia produce cavitación de los segmentos de la médula espinal y los nervios ópticos involucrados, con marcada gliosis y degeneración quística6–8. Las lesiones se encuentran normalmente en las partes centrales de la médula espinal con áreas periféricas: llantas de preservación de la mielina. Se aumenta el número de vasos sanguíneos dentro de la lesión. En las afectaciones agudas activas se ven muchos linfocitos B y pocos CD3+ y CD8+ linfocitos T5. En el caso, con la administración de gadolinio se observó una captación heterogénea del mismo en el interior de la médula a nivel de T5 lo que demuestra el grado de neovascularización.
EpidemiologíaEs más frecuente en mujeres que en hombres (>80%), con un pico de incidencia en la cuarta década9. A diferencia de la EM, que predomina en caucásicos, la NMO afecta con más frecuencia a poblaciones asiáticas y africanas10,11. El paciente no se encuentra en estos parámetros, lo que le convertiría en un caso epidemiológicamente atípico excepto por su edad.
Cuadro clínicoSe caracteriza por ataques recurrentes de neuritis óptica (uni o bilateral) y/o de mielitis transversa extensa, que pueden presentarse de forma simultánea o aislada12. Los síntomas de compromiso del nervio óptico y del cordón espinal pueden presentarse simultáneamente o por periodos separados semanales, mensuales e, incluso, anuales. El compromiso de estos puede ser parcial y en el nervio óptico puede llegar a ser subclínico (sin síntomas visuales, pero con compromiso en los potenciales evocados visuales)12. En la descripción del caso la amaurosis apareció antes que la miopatía transversal y en periodos espaciados de 9 años; la mielopatía es una recaída de su enfermedad que empezó con la neuritis y atrofia óptica.
Criterios de diagnósticoSe han establecido para su diagnóstico 3 condiciones clínicas: neuritis óptica, mielitis aguda y ausencia de síntomas que impliquen compromiso en otro nivel del sistema nervioso central13; en la resonancia magnética, que evidencie lesión longitudinal en T2, con extensión de 3 o más segmentos vertebrales, algo raramente encontrado en la EM; y seropositividad para IgG-NMO, (anti-AQP4)1 es un marcador serológico específico para esta enfermedad2. La identificación del anticuerpo permite diferenciar claramente la NMO de la EM14,15 Es por lo tanto importante como primer paso diagnóstico descartar la EM, como entidad más frecuente en relación con la NMO (tabla 1).
Asociación con otras enfermedadesEsta enfermedad se ha asociado con anticuerpos antifosfolípidos. Se ha descrito una prevalencia del 55% en la presencia de anticuerpos positivos en pacientes con lupus con síntomas de mielitis transversa16,17. En algunos pacientes la primera manifestación del lupus es la NMO18; en el caso no se podría descartar una forma de presentación neurológica del lupus eritematoso sistémico, pero se contó con serología negativa para descartar esta posibilidad. También se ha encontrado relación con infecciones como herpes y Epstein-Barr, e intoxicaciones19,20.
Tratamiento y manejo terapéuticoSe recomiendan tratamientos agresivos con inmunosupresores para reducir la frecuencia de las recaídas.
El tratamiento recomendado en las recaídas es la metilprednisolona, en una dosis de 1g/d, durante 5 días consecutivos, seguida de un tratamiento oral a largo plazo con prednisona, para evitar recaídas. Como alternativa, algunos autores recomiendan el inicio de inmunosupresión con azatioprina (2-3mg/kg/d), asociada a corticosteroides como tratamiento de mantenimiento21. Otra posibilidad terapéutica es el rituximab, en infusión de 375mg/m2, una vez a la semana, durante 4 semanas22.
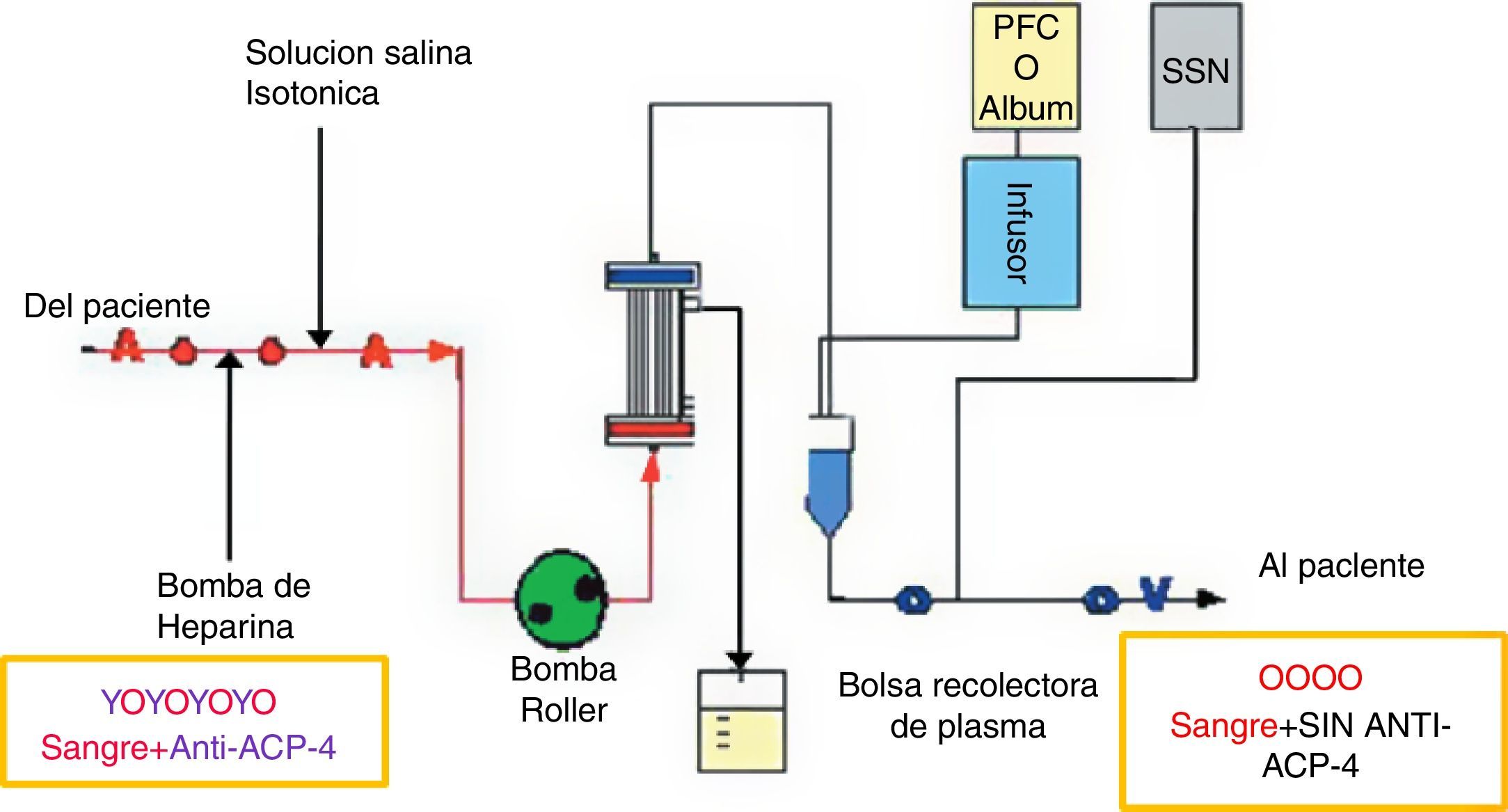
La plasmaféresisConsiste en extraer sangre del cuerpo y procesarla de forma que los glóbulos blancos y rojos se separen del plasma. Las células de la sangre se devuelven luego al paciente sin el plasma, el cual el organismo sustituye rápidamente; en la NMO posee 2 indicaciones: en aquellos pacientes que no responden a los corticosteroides intravenosos (corticoterapia de rescate), y en las recaídas de la enfermedad en un régimen de 55ml/kg en cada recambio, administrado durante 14 días y respetando un día entre cada sesión21. La plasmaféresis elimina eficazmente anticuerpos anti-AQP4 y mejora la discapacidad neurológica en un 50% inmediatamente después del procedimiento, y en un 78% después de 6 meses23; otros estudios recomiendan el uso de inmunosupresión oral y una sesión mínima de plasmaféresis con 4-59 bolsas de plasma para prevenir la recurrencias24. En el momento del diagnóstico se empleó la corticoterapia de rescate, consiguiendo mejoría en cuanto al movimiento; sin embargo, considerando que es una recaída, se indicó la plasmaféresis en 3 sesiones (en la primera se extrajeron 2,331cc de plasma y se restituyeron 2,328cc de plasma; en la segunda se extrajeron 2,320cc y se restituyeron 2,320cc; y en la tercera se extrajeron 2,358cc y se restituyeron 2,320cc). En las evoluciones siguientes a la plasmaféresis fue en donde se observó la mejoría en los momentos del examen motor y de sensibilidad, por lo que el paciente fue dado de alta 5 días después del procedimiento (fig. 5).

Esquema explicativo de la función y el objetivo de plasmaféresis en la eliminación de los anticuerpos contra las AQP4 del plasma. Fuente: Miyamoto y Kusunoki24; elaborado por los autores.
En el curso de la enfermedad puede haber recaídas en el 80% de los casos o ser monofásica, en el 20% restante. La mayoría de los pacientes presenta una recaída en el transcurso del primer año después de su manifestación inicial. El pronóstico de la NMO que no recibe tratamiento es peor que en la EM, más aún cuando las recaídas han sido frecuentes25. Aproximadamente el 30% de los pacientes con NMO mueren al cabo de 5 años después de iniciada la enfermedad, por falla respiratoria, secundaria a lesiones medulares o espinales.
ConclusiónDespués de revisar la bibliografía se espera una nueva recaída en un promedio de entre 3 a 15 años. La terapéutica no va a dar resolución completa a la enfermedad y la historia natural culminaría desfavorablemente con una desmielinización progresiva. Por lo tanto, se indicó las revisiones periódicas a neurología cada 2 meses. Sin embargo, después de administrada la plasmaféresis se notó una importante mejoría en cuanto a que el paciente recuperó sus funciones motoras y de sensibilidad en sus miembros inferiores hasta el punto de que podía sostenerse por sí mismo con el uso de muletas, recuperándose de su estado de postración; en cuanto al aspecto oftalmológico, el paciente mejoró su agudeza visual en el ojo derecho afectado por neuritis en tanto en el izquierdo, afectado por atrofia óptica, no se evidenció ningún cambio.
Responsabilidades éticasProtección de personas y animalesLos autores declaran que para esta investigación no se han realizado experimentos en seres humanos ni en animales.
Confidencialidad de los datosLos autores declaran que en este artículo no aparecen datos de pacientes.
Derecho a la privacidad y consentimiento informadoLos autores declaran que en este artículo no aparecen datos de pacientes.
FinanciamientoLos autores no recibieron patrocinio para llevar a cabo este artículo.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.
