


¿ Introducción
El síndrome de Vogt-Koyanagi-Harada (SVKH), como expresión de inflamación coroidea difusa, en su forma típica se caracteriza por la bilateralidad de un proceso uveomeningítico y panuveítis granulomatosa sin vasculitis retiniana asociada. La forma principal de este síndrome se manifiesta por múltiples desprendimientos serosos retinianos asociados a manifestaciones sistémicas: despigmentación cutánea y de los apéndices y trastornos auditivos y meníngeos.1,2
En su mecanismo fisiopatogénico se mezclan probablemente procesos autoinmunes mediados por clones de células T contra antígenos melanocíticos (proteína de la familia de las tirosinas en células mononucleadas) en sujetos con predisposición genética,3 al parecer desencadenados por una infección viral previa (Epstein-Barr). Parecen estar involucrados los anticuerpos HLA-DR4 y HLD-DRB1.4,5
Se describen cuatro periodos clínicos. La fase prodrómica, de tres a cinco días de duración; que se caracteriza por la presencia de síntomas neurológicos y disacusia. En la uveítica se presentan los síntomas oculares: visión borrosa como síntoma cardinal (70% de los casos) y signos de panuveítis bilateral. El primer signo detectable al examen físico lo constituye el desprendimiento seroso de la retina, acompañado de edema del nervio óptico por engrosamiento coroideo. El periodo crónico/ convaleciente se caracteriza por una imagen fundoscópica en "puesta de sol" (más frecuente en asiáticos), nódulos de Dalen-Fuchs, signo de Sugiura, focos de hiperpigmentación, zonas de atrofia focal periféricas del epitelio pigmentario de la retina (EPR) y signos tegumentarios (menos frecuentes en caucásicos).
En la recurrencia, las formas de presentación más frecuentes son: uveítis anterior granulomatosa refractaria al tratamiento esteroideo, rara la toma posterior, presencia de nódulos en el iris, atrofia del mismo, catarata, glaucoma, alteraciones del EPR y neovascularización coroidea.1,6
Los criterios diagnósticos de VKH establecen tres formas clínicas, en dependencia de la presencia o ausencia de los diferentes criterios:1 se considera VKH completo (todos presentes), VKH incompleto (criterios uno a tres obligatorios y cuatro o cinco presentes) y VKH probable (condición ocular aislada, presentes de uno a tres criterios):
1. No antecedentes personales de trauma o cirugía ocular previos al cuadro uveal.
2. No evidencias clínicas o de laboratorio de otras causas de uveítis.
3. Condición ocular bilateral.
4. Hallazgos neurológicos o auditivos, manifiestos en etapas iniciales o bien documentados: meningismo dado por malestar, fiebre, cefalea, náuseas, dolor abdominal, rigidez de nuca o varios de los mismos, tinnitus o pleocitosis del LCR.
5. Hallazgos cutáneos o tegumentarios que no anteceden inicio de las alteraciones neurológicas: alopecia, poliosis o vitiligo simétrico.
El diagnóstico se realiza mediante el examen ocular completo (que incluye biomicroscopía del segmento anterior y posterior, y oftalmoscopía binocular indirecta). Se indica angiografía con fluoresceína (AFR) o con verde indocianina, pruebas de laboratorio (hemograma completo, VDRL, proteína C reactiva, factor reumatoide e IgG), estudios
de imagen y del líquido cefalorraquídeo (LCR) y determinación de HLA si es posible.7 La ecografía, la tomografía de coherencia óptica y la biomicroscopía ultrasónica, pueden resultar útiles en algunos casos y para la evolución clínica.8
La primera línea terapéutica la constituyen los corticoesteroides sistémicos, en forma de pulsos o en dosis fraccionadas por vía intravenosa u oral (de acuerdo a severidad, riesgo, etc.), con reducción progresiva personalizada entre seis meses y un año. Otra opción, si no es posible el uso de los corticoesteroides, son los inmunosupresores (ciclofosfamida, clorambucil, azatioprina). Como terapia de apoyo se emplean esteroides tópicos y ciclopléjicos.9,10
Se presenta un caso en fase uveítica clásica, sin síntomas neurológicos previos al cuadro ocular y con un antecedente poco común.
¿ Presentación del caso
Se trata de una paciente de 43 años, piel blanca, que acude al Centro por el Servicio de Urgencia. La paciente refirió rápida y progresiva disminución de la agudeza visual (AV) de ambos ojos, principalmente del izquierdo, acompañada de fotofobia, dolor y ojo rojo, de más o menos cinco días de evolución. El cuadro había sido interpretado como un proceso infeccioso conjuntival que, al no tener resolución y empeorar la sintomatología, la motivaron a acudir al Centro. No se recogieron otros síntomas y como antecedentes patológicos personales, hepatitis C tres años atrás, tratada con interferón α-2 recombinante durante 12 semanas, evolutivamente estable desde entonces y con marcador de carga viral negativo. Como antecedentes familiares, solo refirió hipertensión arterial.
Al examen oftalmológico se recogieron los siguientes datos:
Hiperemia cilioconjuntival marcada AO.
Segmento anterior: OD: precipitados retroqueráticos gruesos, sobre todo en mitad inferior, celularidad del acuoso 2+/ flare 2+, OI: precipitados retroqueráticos finos y gruesos con distribución global, celularidad del acuoso 3+/flare 3+.
Medios: OD: celularidad vítrea 2+, OI: celularidad vítrea 2-3+.
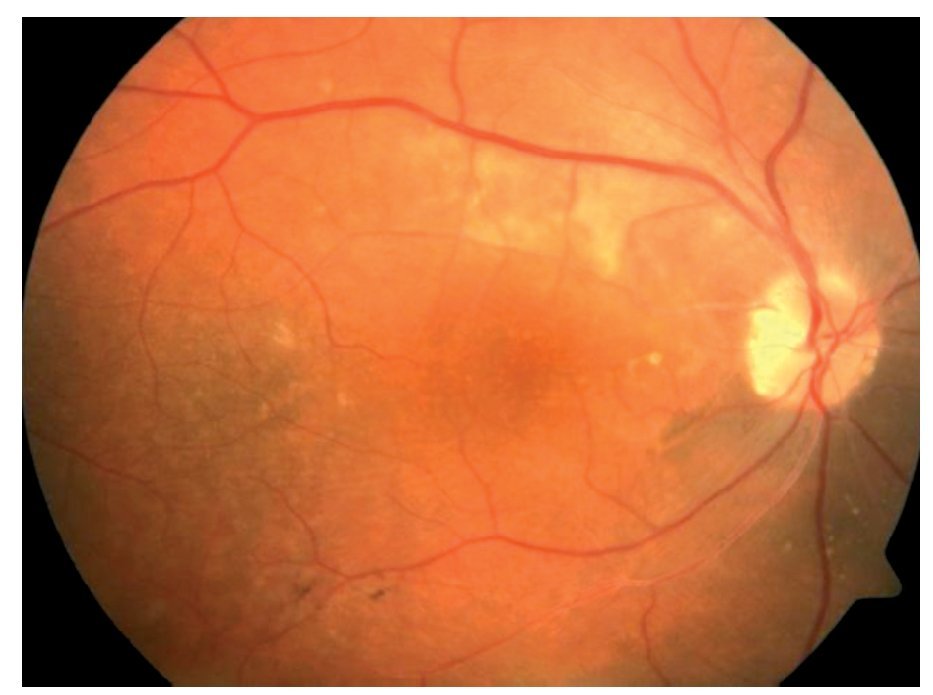
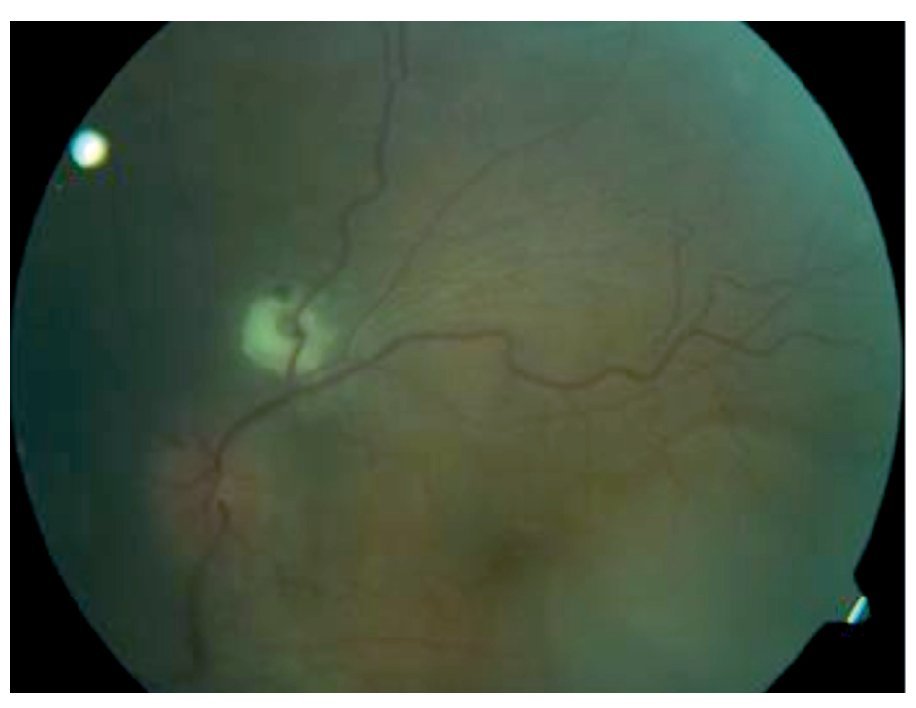
Fondo de ojo: OD: papila ligeramente edematosa con borde nasal borroso, desprendimiento seroso en arcadas superior e inferior, disminución de brillo foveal, vasos sin alteraciones, OI: papila edematosa, desprendimiento seroso en arcadas superior y temporoinferior a la fóvea, vasos tortuosos (Figuras 1 y 2).
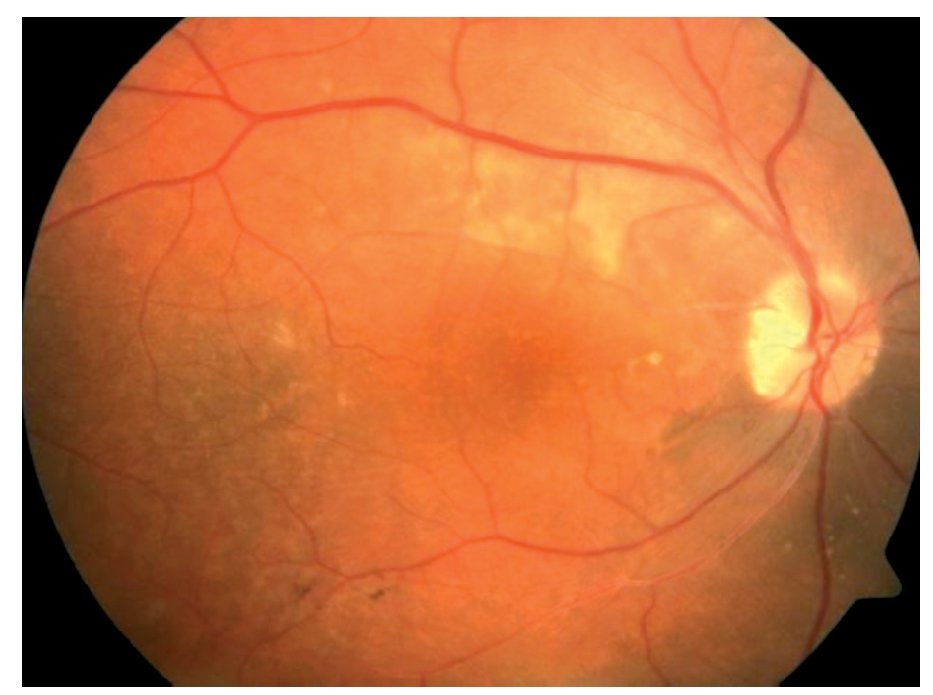
¿ Figura 1. Imagen fundoscópica de la paciente.
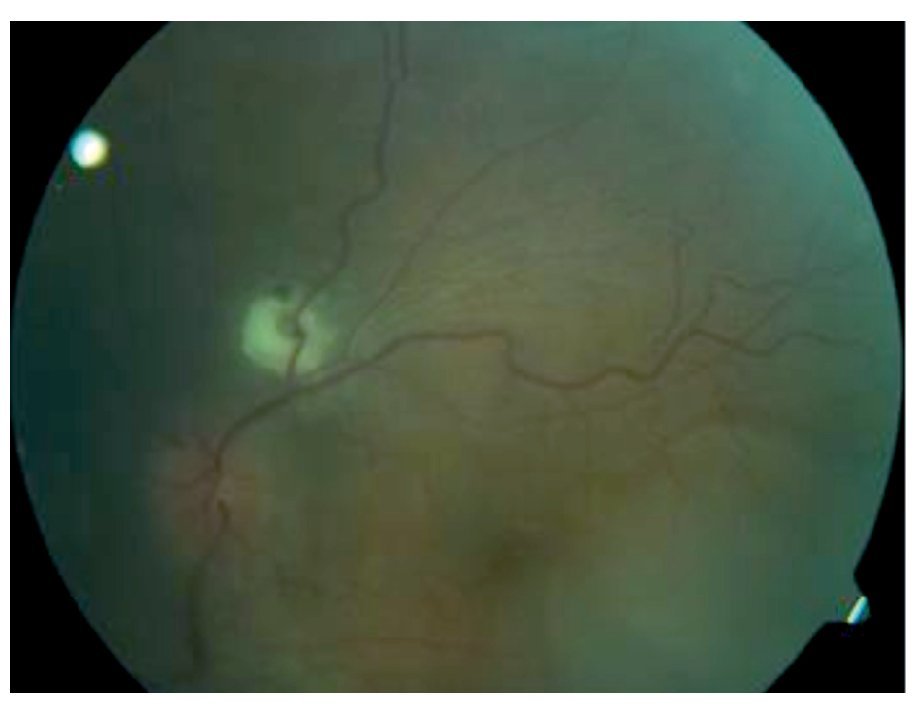
¿ Figura 2. Imagen fundoscópica de la paciente.
AV (Snellen -cc-): OD: 0.5 +1.00, OI: 0.3 +1.00.
PIO (Goldmann): OD: 12 mmHg, OI: 8 mmHg (12:30 h).
El examen físico general no arrojó alteraciones.
Se decidió ingresar a la paciente, se proporcionó el tratamiento inicial y se inició su estudio. Se impuso tratamiento inicial con antiinflamatorios esteroideos: 1 g/300 mL de solución salina isotónica por tres días. Al cuarto día, prednisolona 1 mg/kg/día, con reducción progresiva a razón de 5 mg semanales. Desde el punto de vista tópico, se indicó homatropina 2% y dexametasona.
Investigaciones:
Leucograma: leucocitosis ligera con predominio de linfocitos.
Eritrosedimentación: 12 mm/h.
Pruebas serológicas para sífilis, tuberculosis, VIH, toxoplasmosis: negativas.
Ecografía hepática: normal.
Rayos X de tórax: normal.
LCR: pleocitosis ligera con predominio linfocitario.
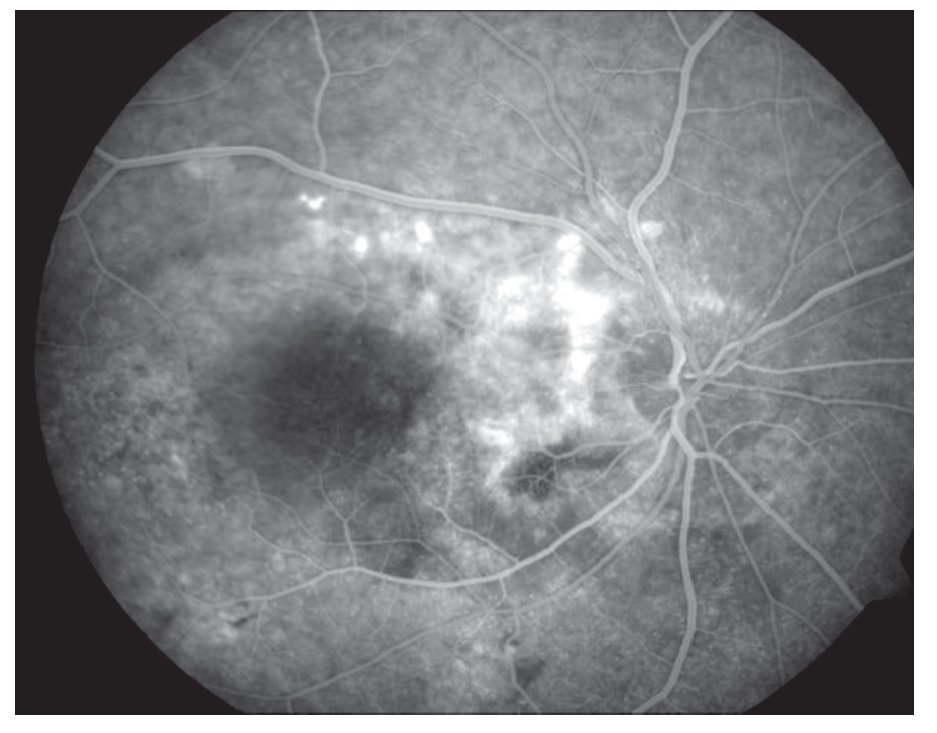
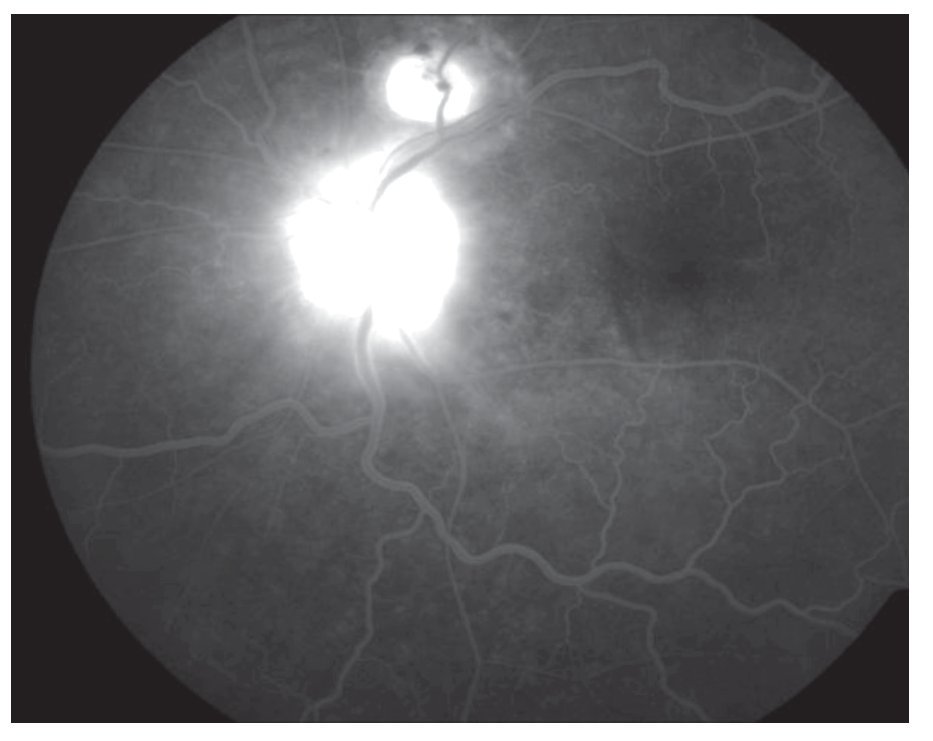
AFR: OD: hiperfluorescencia entre las arcadas por acúmulo de fluoresceína en el espacio subretinal en correspondencia con el desprendimiento seroso y defectos en ventana en áreas atróficas periféricas. OI: hiperfluorescencia en la arcada superior en correspondencia con el desprendimiento seroso y del nervio óptico (Figuras 3 y 4).
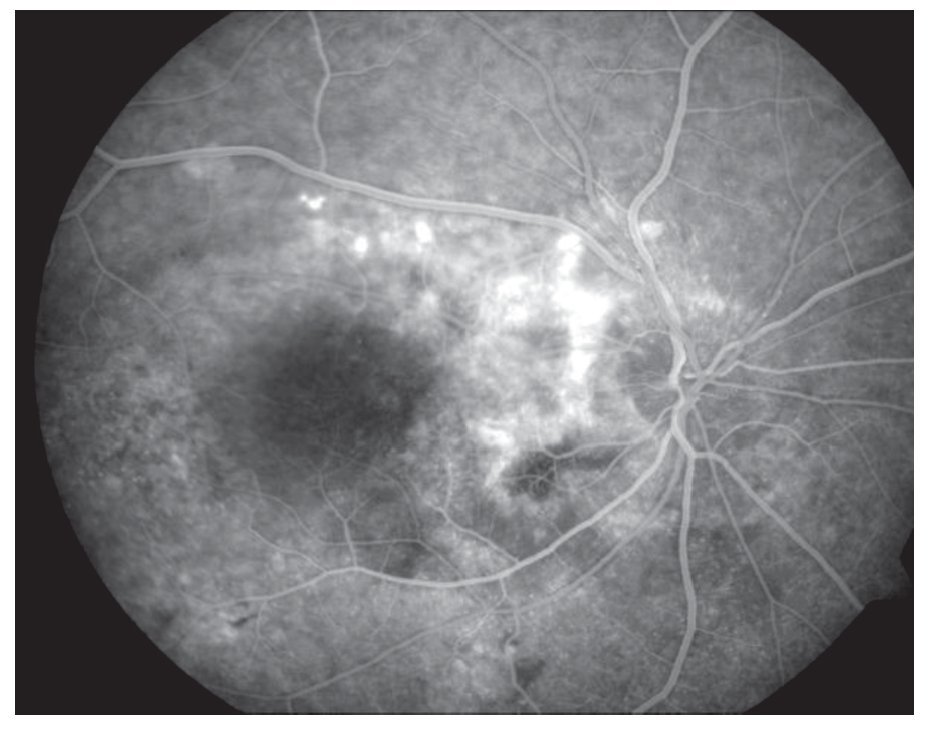
¿ Figura 3. Imagen angiográfica (AFR) de la paciente.
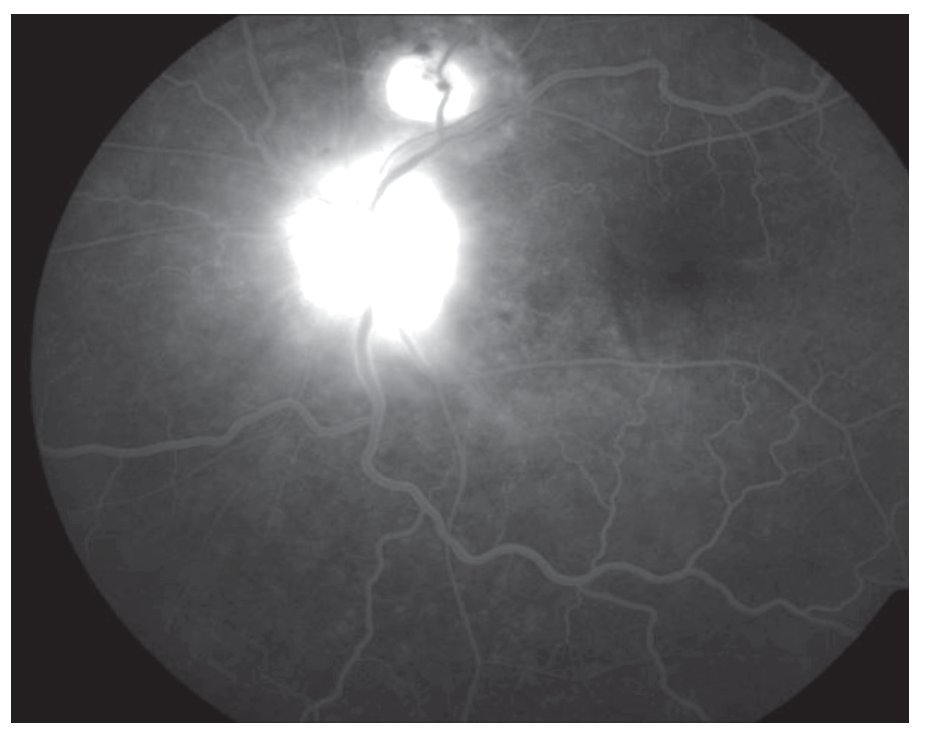
¿ Figura 4. Imagen angiográfica (AFR) de la paciente.
La paciente evolucionó favorablemente y después de un año de evolución, no se ha presentado recurrencia.
¿ Discusión
Después de concluido el estudio, y dada la evolución del caso, se concluyó como una probable enfermedad de VKH por las características del cuadro oftalmológico bilateral, la ausencia de manifestaciones extra-oculares, de antecedentes de trauma o cirugía ocular previos al cuadro uveal y la no evidencia clínica o de laboratorio de otras causas de uveítis.1,2
El posible mecanismo fisiopatológico en este caso pudo deberse a una reacción autoinmune desencadenada por la infección vírica previa (hepatitis C) con uso terapéutico de interferón α-2 recombinante. En 2006, un estudio publicaba que el interferón α-2b había sido relacionado con la posible aparición de VKH en pacientes con hepatitis C.11
Se realizó el diagnóstico diferencial con la oftalmía simpática por la ausencia de antecedente de trauma penetrante ocular; con la escleritis posterior, más frecuente en mujeres y bilateral desde el inicio, dada la ausencia de antecedente de artritis reumatoide; con el linfoma primario por la ausencia de manifestaciones neurológicas, por el tipo de manifestaciones fundoscópicas y las características de la angiografía y con la sarcoidosis por la presencia del desprendimiento seroso de la retina y la ausencia de vasculitis retinal. Con otras enfermedades menos frecuentes, como el síndrome de efusión uveal, la coroidopatía lúpica, la hiperplasia melanocítica y la borreliosis ocular (Enfermedad de Lyme) por la ausencia de antecedentes y las características clínicas y angiográficas.7,12
Correspondencia: Dra. Idalia Triana Casado.
Calzada del Cerro N°1551 esquina a Domínguez, Cerro. La Habana, Cuba.
Teléfono: 877 6358.
Correo electrónico: idalia.triana@infomed.sld.cu.
