


El síndrome de Axenfeld-Rieger es una entidad de origen genético que se caracteriza por diversos hallazgos oculares. En este artículo se presenta el caso de una paciente de 54 años de edad que es diagnosticada con este síndrome y en cuya exploración se hace evidente la presencia de catarata de manera bilateral, así como alteraciones en la movilidad ocular. Se describen los hallazgos clínicos, ecográficos y por ultrabiomicroscopia, y además se hace una revisión de la literatura sobre el tema.
The Axenfeld-Rieger syndrome is a genetic entity characterized by various ocular findings. In this article we present the case of a 54-year-old patient who is diagnosed with this syndrome and that within its examination the presence of bilateral cataracts and ocular motility abnormalities are evident. The clinical, echographic and ultrabiomicroscopy findings are described, and also a review of the literature is included.
El síndrome de Axenfeld-Rieger es una enfermedad de origen genético, que afecta múltiples órganos que tienen en común su origen en la cresta neural durante el desarrollo embrionario y fetal1,2. Dentro de las múltiples malformaciones destacan las craneofaciales, cardiovasculares, musculoesqueléticas, del oído y de la glándula hipófisis. El cuadro clínico ocular característico es el embriotoxón posterior, sinequias anteriores periféricas, atrofia marcada del iris, seudopolicoria y corectopia, además de un riesgo elevado de glaucoma3.
Presentamos el caso de una paciente con síndrome de Axenfeld-Rieger en asociación con una endotropia progresiva por alta miopía, así como los hallazgos encontrados por ecografía y en la ultrabiomicroscopia (UBM).
Presentación del casoPaciente femenino de 54 años que acude a consulta oftalmológica por presentar visión borrosa de ojo derecho (OD) de 6 meses de evolución que ha sido progresiva, además de desviación ocular de su ojo izquierdo (OI) de 2 años de evolución.
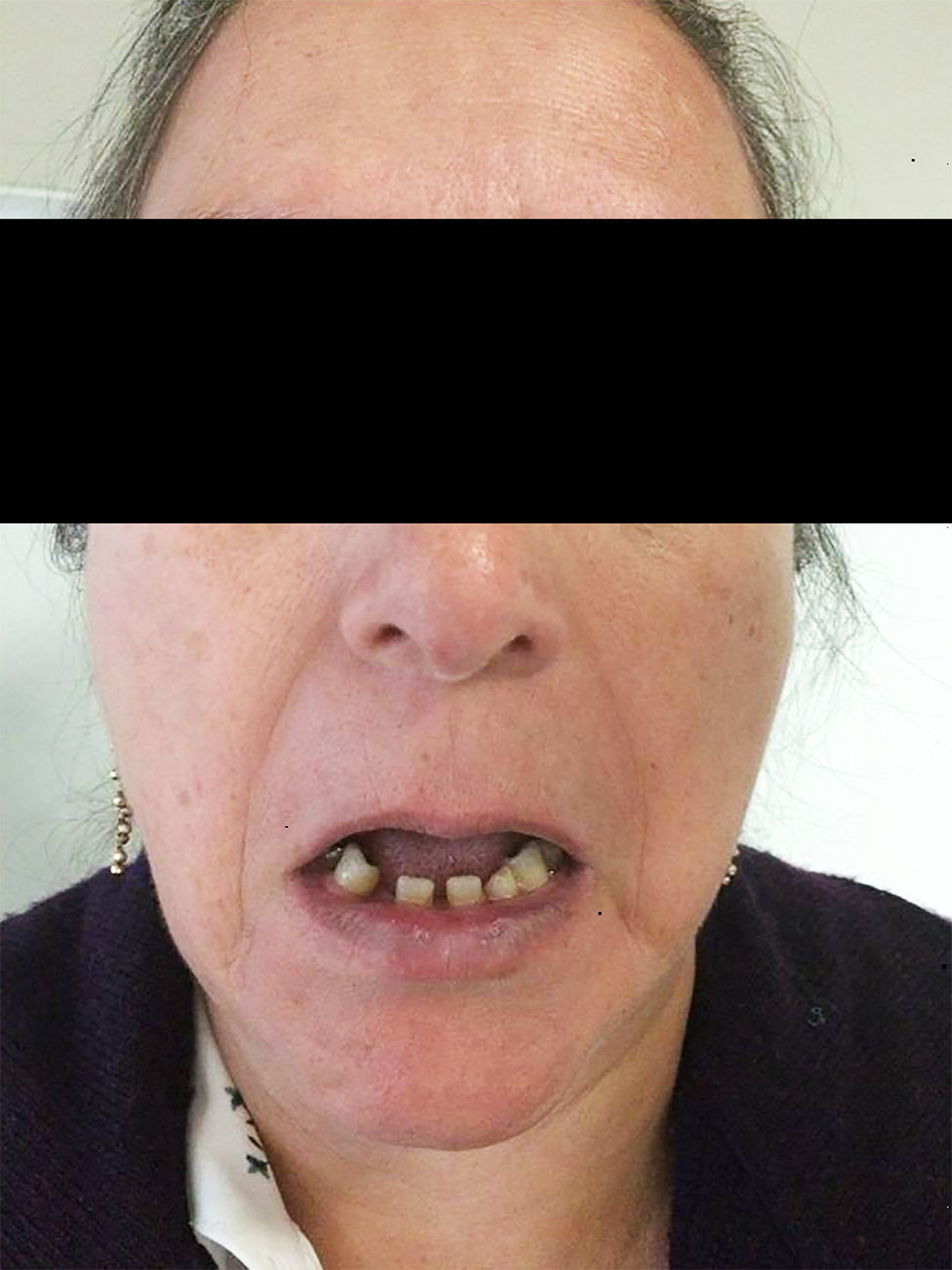
Es originaria del Distrito Federal y ama de casa. Dentro de sus antecedentes heredofamiliares, su madre e hijo con diagnóstico de glaucoma, padre diagnosticado con síndrome de Rieger y un hijo con coloboma aparentemente del iris. Usa lentes de contacto blandos desde los 16 años y aéreos desde los 18 años, niega cirugías oftalmológicas. Refiere anomalía dental desde el nacimiento, tratado con prótesis (fig. 1), así como antecedente de hernioplastia umbilical.

A la exploración inicial se midió la agudeza visual de OD en 20/300 que con estenopeico mejoraba a 20/200, y en el OI 20/200 que no mejoraba. La retinoscopia no fue satisfactoria por opacidad de medios. La presión intraocular se tomó con tonómetro de aplanación en 15 y 20mm Hg respectivamente.
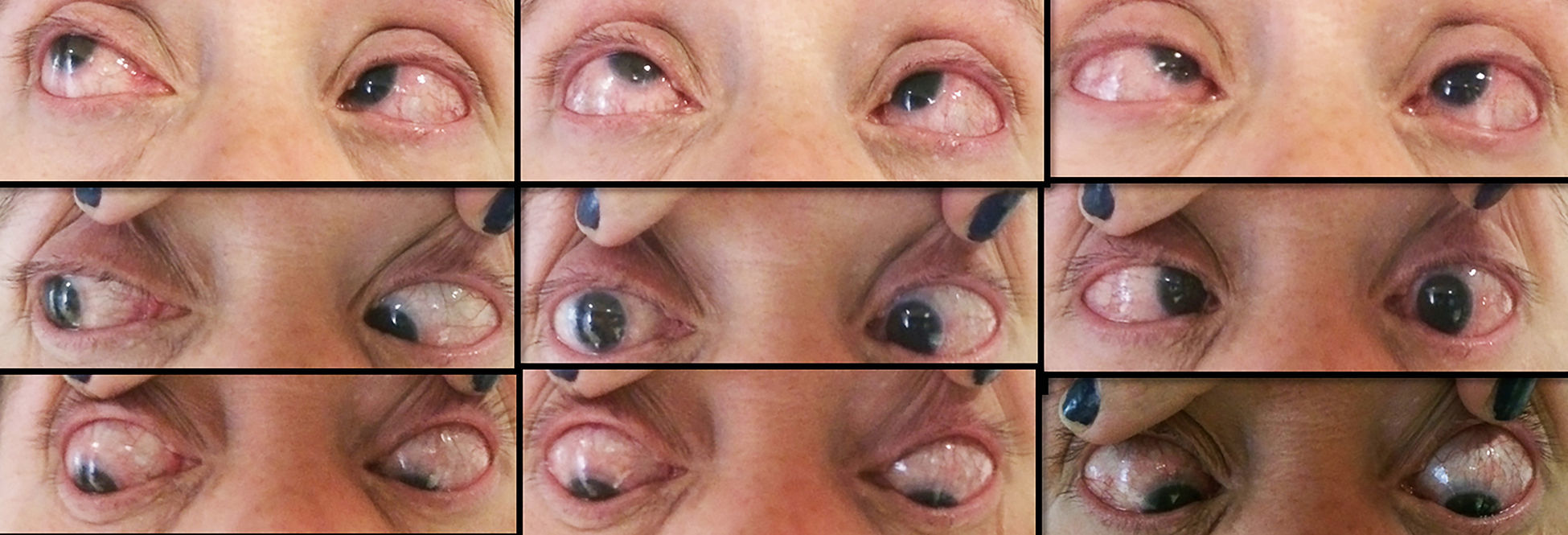
A la exploración estrabológica se encontró una seudoptosis con posición compensadora de la cabeza elevando el mentón, siendo el ojo preferente el derecho. En la mirada al frente una endotropia de 50dioptrías prismáticas (DP) con hipertropia de 15DP con limitación a la levoversión de −5 OI y −1 a la dextroversión del mismo ojo, además de limitación a la supra e infraversión de −2 en OI (fig. 2). Presentó incomitancia a la lateroversión derecha por hipotropia del ojo en aducción. Se realizó prueba de ducción forzada pasiva de OI hacia arriba y hacia fuera, siendo positivas 3+. Se realizó el diagnóstico clínico de endotropia progresiva por alta miopía.

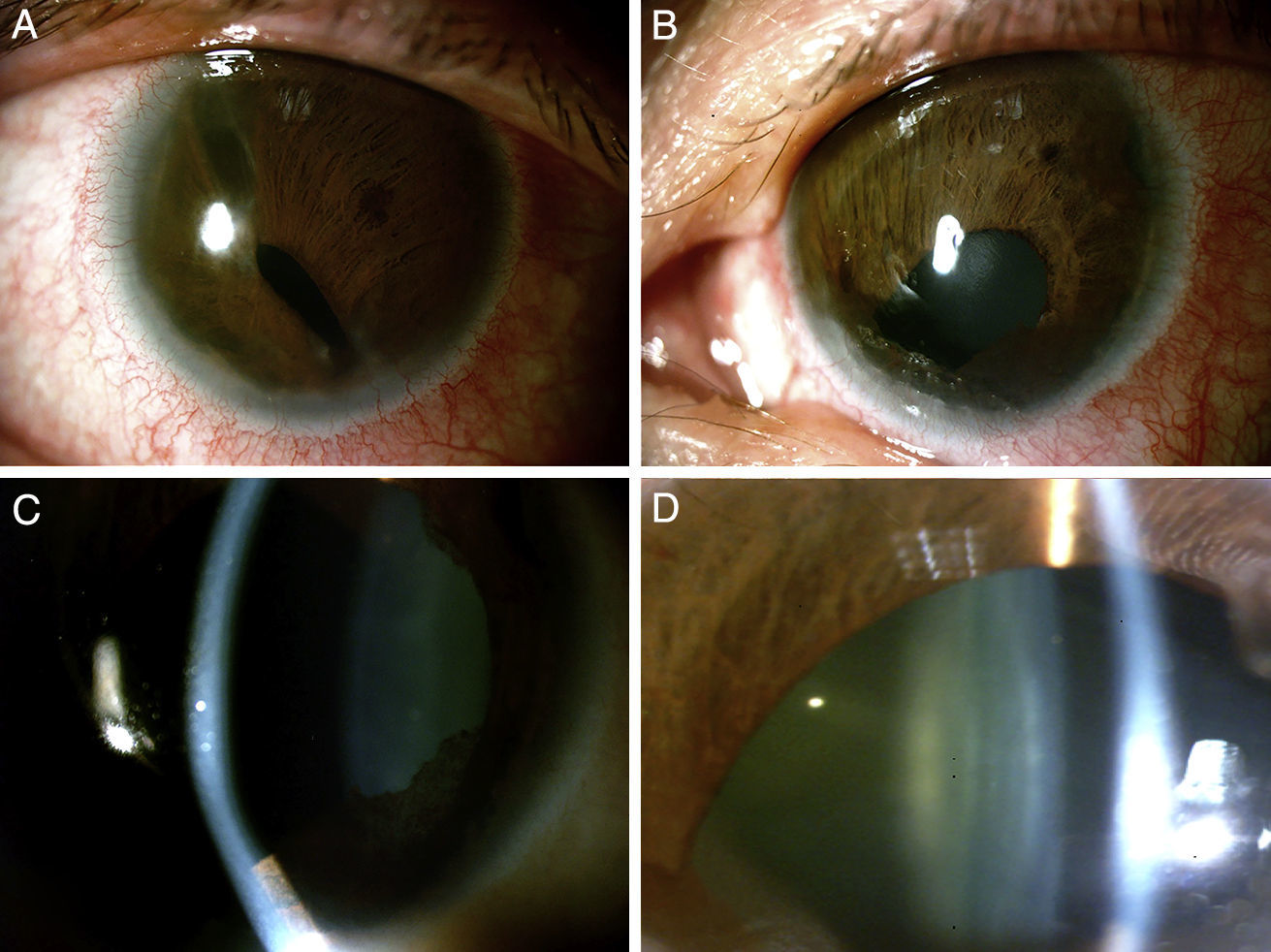
En la biomicroscopia del segmento anterior, en OD se encontró conjuntivas con hiperemia leve difusa, córnea clara con embriotoxón posterior 360 grados, con cámara anterior formada, iris con presencia de sinequias anteriores periféricas, seudopolicoria y corectopia, cristalino con opacidad (fig. 3). En el OI, el segmento anterior con mismos hallazgos, pero corectopia con pupila hacia sector inferior (fig. 3). En la gonioscopia de ambos ojos se observó apertura en 180 grados y el resto con sinequias anteriores periféricas a la línea de Schwalbe (fig. 4). Bajo dilatación farmacológica se encontró en OD un fondo coroideo, y el resto no era valorable por opacidad de medios. En OI la fundoscopia mostró un fondo coroideo en una retina aplicada con una papila naranja de bordes bien definidos, de inserción oblicua, con atrofia beta y excavación aproximada del 45%, con anillo neurorretiniano conservado y mácula sin brillo foveolar.


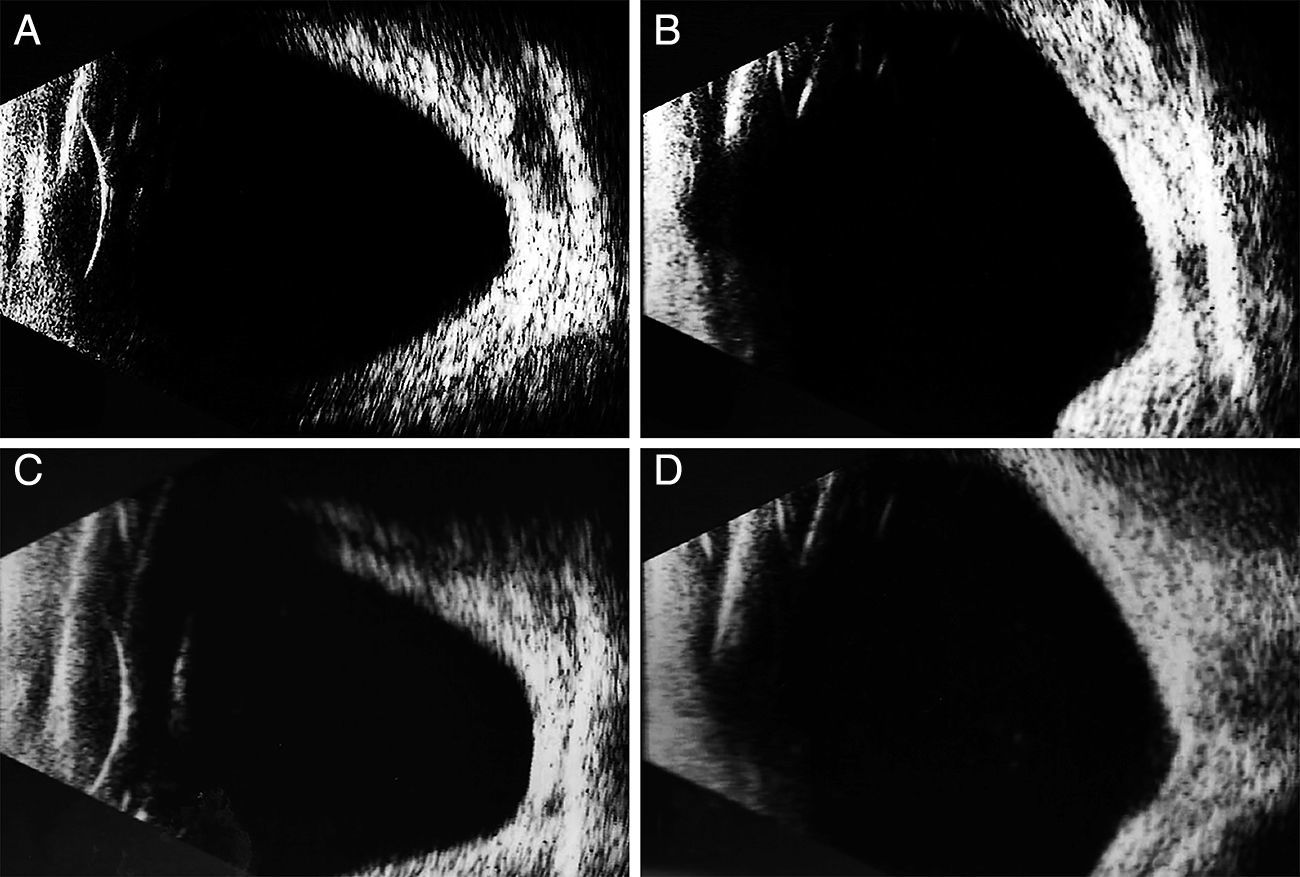
Se realizó ecografía modo A biométrico, modo B con sonda de 10MHz y UBM con sonda de 50MHz de ambos ojos. Se encontró una longitud axial grande en ambos ojos, con 31.59mm en OD y 30.93mm en el OI. En el ultrasonido modo B se observó que ambos ojos eran fáquicos, con discretas opacidades en cristalino sugestivas de catarata, el vítreo tenía mínimas opacidades y la hialoides posterior se observaba parcialmente desprendida, sin tracciones vitreorretinianas; la retina y la coroides estaban aplicadas y la excavación del nervio óptico era poco valorable por presencia de estafiloma posterior, con involucro macular (fig. 5).

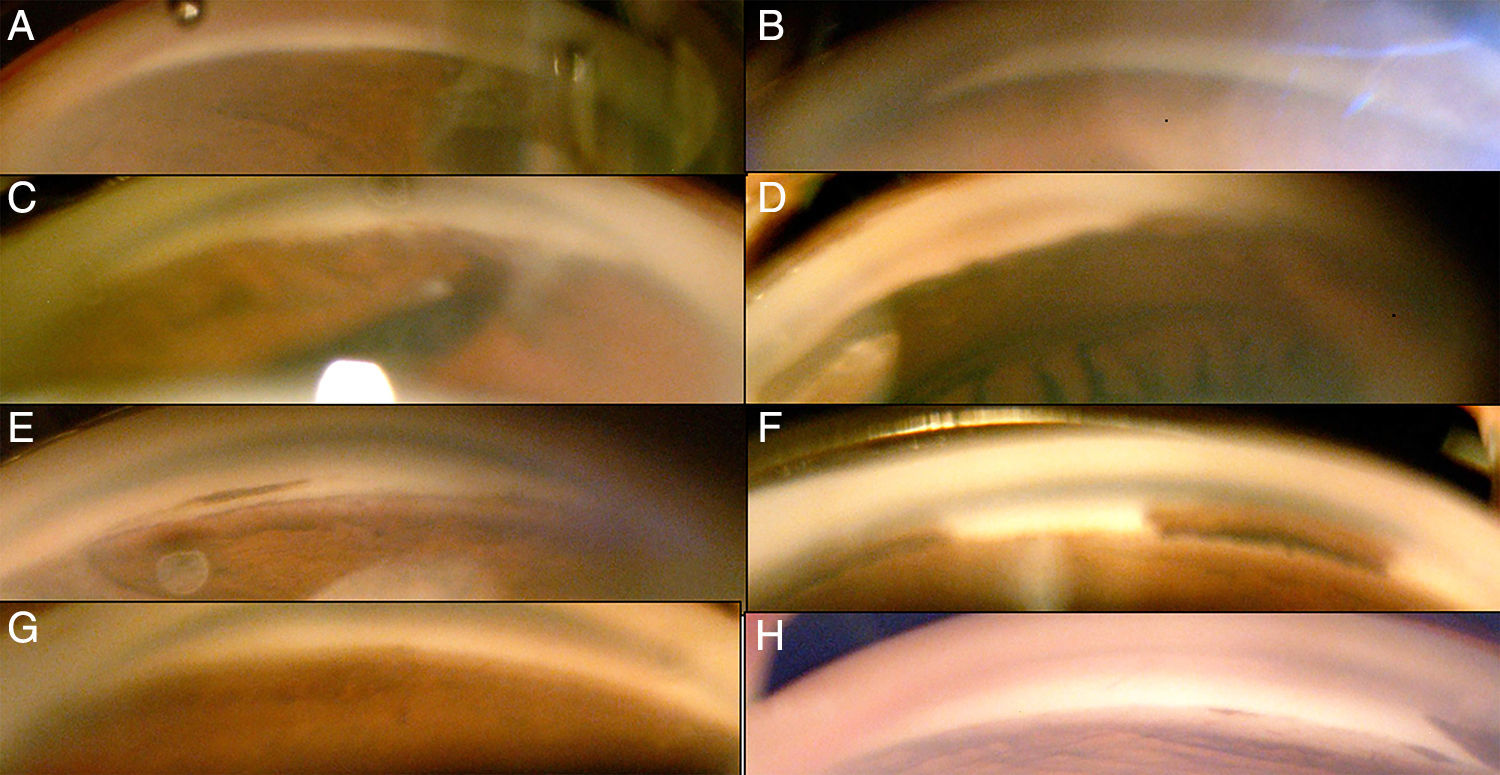
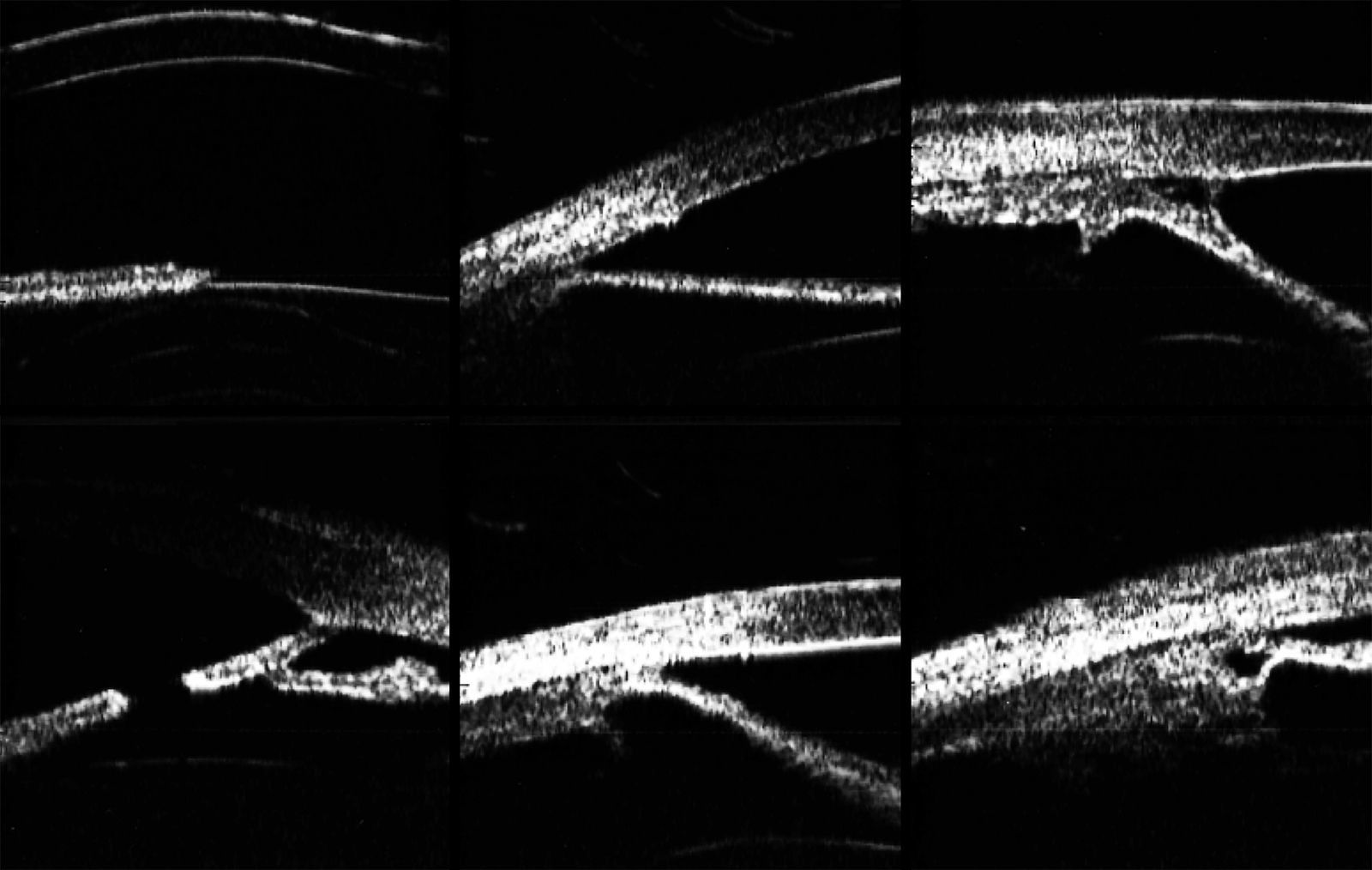
En la UBM se documentó una córnea uniforme y con grosor central de 561μm en OD y 487μm en OI, con la cámara anterior formada pero estrecha, midiendo 1.86mm en OD y 2.50mm en OI. El iris se observaba delgado, atrófico y plano, excepto en su periferia donde tenía su inserción más anterior además de presentar adherencias hacia la córnea, que corresponden a las adherencias a una línea de Schwalbe prominente. El ángulo en los sectores donde tenía las sinequias era cerrado, pero en los sectores libres estaba abierto, a pesar de la inserción más anterior del iris. Debido a esta configuración, el surco ciliar era amplio, con un cuerpo ciliar aplicado pero pequeño y atrófico, así como ecos en cristalino sugerentes de catarata (fig. 6).

El plan de tratamiento de la paciente consiste en realizar cirugía de facoemulsificación de catarata con implante de lente intraocular, con posterior manejo por el departamento de estrabismo.
DiscusiónLas anomalías del desarrollo del segmento anterior en ocasiones involucran la córnea, el iris, el ángulo iridocorneal y el cristalino. Previamente se consideraba que el síndrome de Axenfeld-Rieger formaba parte de los llamados síndromes de segmentación de la cámara anterior o también llamados disgenesias mesodérmicas2,3. La anomalía de Axenfeld se caracteriza por la presencia de un embriotoxón posterior con bandas de iris insertadas, y cuando se agrega glaucoma se conoce como síndrome de Axenfeld. La anomalía de Rieger consiste en la anomalía de Axenfeld agregándose la hipoplasia del estroma anterior del iris, dando como consecuencia corectopia y seudopolicoria. El síndrome de Rieger era descrito como la anomalía del mismo nombre más anormalidades sistémicas como hipoplasia maxilar y hernia umbilical. Finalmente, la anomalía de Peters consiste en un defecto en la cara posterior de la córnea, con opacidad del estroma en esta zona y bandas de iris o incluso de cristalino adheridas al defecto4. Recientemente se ha considerado que todas estas anormalidades forman parte en realidad del mismo síndrome, siendo aceptado el término general de síndrome de Axenfeld-Rieger para referirse a todo este espectro de la enfermedad2.
Este síndrome se hereda de manera autosómica dominante, y son 2 los genes que se han implicado de manera más importante, uno es PITX2 en 4q25, presente en el 10-60% de los enfermos, asociado principalmente a alteraciones sistémicas como las dentales y umbilicales5–7. El otro gen responsable es FOXC1 localizado en 6q25, presente en el 50% de los casos y es el que se ha asociado con las manifestaciones oculares, sobre todo el glaucoma5,7,8.
Existe una amplia lista de manifestaciones sistémicas en estos pacientes. Dentro de la afección cardiovascular destacan la persistencia del tronco arterioso, anomalías en las válvulas cardiacas y, de especial importancia, la afección del septum interauricular que conlleva un mayor riesgo de accidentes cerebrovasculares de tipo isquémico y que debe considerarse si se instaura tratamiento antiglaucomatoso2. Dentro de las malformaciones craneofaciales se encuentran la hipoplasia mediofacial, raíz nasal amplia, hipoplasia del maxilar que puede originar prognatismo, labio superior delgado e inferior evertido. Las alteraciones dentales son muy características con microdontia, hipodontia u oligodontia. En cuanto al aspecto musculoesquelético, los afectados pueden tener aplanamiento de las epífisis del húmero y del fémur, vértebras cuadradas y pie zambo. Otras anormalidades que se pueden encontrar son sordera neurosensorial, piel redundante periumbilical, hernia umbilical o inguinal, estenosis anal, hidrocefalia o síndrome de la silla turca vacía2.
Dentro de las manifestaciones oculares, es frecuente el embriotoxón posterior, que se trata de una línea de Schwalbe prominente y desplazada anteriormente, de 0.5-2mm del limbo esclerocorneal, involucrando incluso los 360 grados de la cornea9. Es importante señalar que hasta el 8-15% de las personas sanas pueden tener un embriotoxón posterior, y que algunos casos de síndrome de Axenfeld-Rieger no lo presentan10. Las alteraciones angulares aumentan el riesgo de glaucoma en estos pacientes; se observan las características sinequias del iris al embriotoxón. Las anormalidades en el iris son las características más notables, y puede ser desde una atrofia ligera hasta casi ausencia del iris, lo cual se manifiesta como corectopia, seudopolicoria o más raramente ectropión uveal11. El glaucoma se presenta en alrededor del 50% de los casos. Se han descrito otros hallazgos oculares que incluyen desprendimiento de retina12, microesferofaquia13, hipertelorismo, esclerocórnea, membrana pupilar persistente, queratocono posterior, microftalmos o ambliopía. Debido a que uno de los genes implicados, PITX2, tiene que ver con el desarrollo de los músculos extraoculares, también pueden padecer estrabismo por anomalías en las inserciones musculares, o incluso hipoplasia/agenesia de estos14,15. Hasta nuestro conocimiento, este es el primer caso de Axenfeld-Rieger asociado a una MAPE.
Los estudios de imagen del segmento anterior del ojo pueden ser útiles como apoyo en el estudio de la desorganización de las estructuras anatómicas, teniendo disponibles para este fin principalmente la UBM y la tomografía de coherencia óptica. La tomografía de coherencia óptica del segmento anterior puede mostrar hallazgos similares, además de que con la tecnología de alta resolución se puede observar la membrana endotelial que recubre el iris y el ángulo, y que se supone es la responsable del glaucoma y de la atrofia del iris, con la ventaja de que es un estudio de no contacto16. La desventaja inherente a la tecnología óptica que se emplea es que no se puede visualizar más allá de tejidos pigmentados, como el iris. En un reporte de UBM de un caso de Axenfeld-Rieger se documentó un iris plano, delgado y atrófico, un ángulo con sinequias entre el iris y una línea de Schwalbe prominente, con una inserción más anterior del primero, y debido a esto un sulcus amplio, además de un cuerpo ciliar pequeño y atrófico, visualizándose además un cristalino intumescente17. Debe considerarse la UBM como el estudio de elección cuando se quiere visualizar el segmento anterior completo, ya que es posible visualizar más allá del pigmento del iris, siendo especialmente útil para afección retroiridiana, el surco ciliar, los procesos ciliares, la zónula, parte del cristalino e incluso áreas de la pars plana y de retina muy periférica18,19.
Son diversas las patologías que se deben tomar en cuenta como diagnóstico diferencial, e incluyen el síndrome iridocorneal endotelial, la anomalía de Peters, la distrofia polimorfa posterior, displasia oculodentodigital, iridogoniodisgenesia, aniridia, coloboma de iris, ectopia lentis et pupilae, entre otros.
Para el tratamiento del glaucoma en este síndrome, respecto al manejo tópico se deben emplear supresores de la secreción del humor acuoso, teniendo especial precaución en el uso de medicamentos tópicos adrenérgicos. En el manejo quirúrgico se prefiere realizar un procedimiento de drenaje como una trabeculectomía con mitomicina C o bien un dispositivo valvular; el tratamiento con láser no resulta efectivo. Siempre que se sospeche el síndrome de Axenfeld-Rieger debe evaluarse a los familiares. Se requiere de un manejo multidisciplinario con relevancia en el cardiólogo, por el riesgo de muerte que tienen los pacientes debido a accidentes cerebrovasculares.
ConclusionesEl síndrome de Axenfeld-Rieger se caracteriza por tener una banda del iris hacia la línea de Schwalbe desplazada anteriormente e hipoplasia del iris, con afección congénita bilateral, esporádica o autosómica dominante, con la posibilidad de aparición de glaucoma de forma temprana o tardía. Entre las alteraciones oculares asociadas destacan glaucoma y, en ocasiones, estrabismo. El diagnóstico es clínico, siendo de especial ayuda los antecedentes familiares y la asesoría genética, con una adecuada exploración oftalmológica, principalmente en el segmento anterior. La UBM se prefiere para visualizar de mejor manera los diversos cambios en el segmento anterior característicos del síndrome.
Responsabilidades éticasProtección de personas y animalesLos autores declaran que para esta investigación no se han realizado experimentos en seres humanos ni en animales.
Confidencialidad de los datosLos autores declaran que en este artículo no aparecen datos de pacientes.
Derecho a la privacidad y consentimiento informadoLos autores declaran que en este artículo no aparecen datos de pacientes.
FinanciamientoLos autores no recibieron patrocinio para llevar a cabo este artículo.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.
