


La disgenesia gonadal es una forma rara de hiperplasia suprarrenal congénita con defecto en la biosíntesis del cortisol y esteroides sexuales, que resulta en exceso de mineralocorticoides, hipertensión hipokalémica y anormalidades sexuales como seudohermafroditismo en hombres e infantilismo sexual en mujeres.1-4 La incidencia es de 1:50 000 a 1:100 000 recién nacidos.4-8
Tiene un patrón autosómico recesivo, correspondiendo a 1% de los casos, y es causado por mutaciones en el gen de la citocromo P450c17(CYP17), que se encuentra en el cromosoma 10q24.3 y está constituido por ocho exones, expresándose en la corteza adrenal, ovario y testículos; es mediador de la actividad de la 17 a hidroxilasa y la 17,20-Liasa.3,6,7,9-11 Se describió la primera vez en 1966 por Biglieri y colaboradores en una mujer genotípica, con hipertensión, hipokalemia y retardo de la pubertad. En 1970, se publicó la afección de un hombre genotípico, presentándose con pseudohermafroditismo.5,6,9 Se han descrito poco más de 150 casos en la bibliografía.9
El objetivo de este trabajo es informar sobre dos casos de disgenesia gonadal secundaria a deficiencia de 17a-hidroxilasa, su cuadro clínico, el método para su diagnóstico y el manejo utilizado.
¿ PRESENTACIÓN DE CASO 1
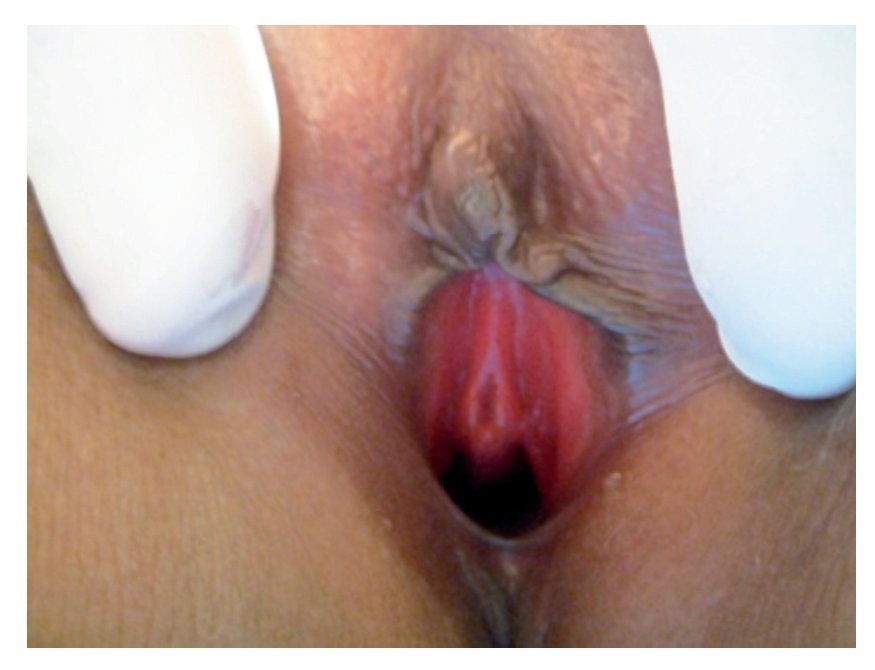
Mujer de 18 años, sin antecedentes de importancia. Amenorrea primaria: G-0. Nunca había tenido actividad sexual. Inició su padecimiento tres días previos a la valoración inicial, con presencia de sensación de debilidad en miembros pélvicos de manera bilateral con pesantez de los mismos, dolor punzante que aumentaba con la actividad física imposibilitando deambulación principalmente en región de muslos de manera bilateral, además tinitus, cefalea de tipo punzante de predominio frontal y presencia de epistaxis en dos ocasiones. A la exploración con TA 170/80 mmHg. Cardiopulmonar sin compromiso, ausencia de desarrollo de mamas Tanner I, sin presencia de vello axilar. Abdomen sin alteraciones, genitales externos con ausencia de vello púbico (Imagen 1); no se realizó tacto vaginal (paciente núbil). Extremidades inferiores con presencia de aumento de tono muscular, con fuerza disminuida, dolor a la palpación de grupos musculares, reflejos osteotendinosos conservados, Babinsky negativo, con movimientos de articulaciones conservados. Exámenes de laboratorio dentro de los parámetros normales; sólo la DHL: 1213 UI, potasio: 2.1 mEq/L; Gasometría Arterial con pH 7.6, PCO2: 36.2, PO2: 58.5, HCO3: 36.6, SO2: 93.3%. EGO con densidad de 1.001, 12-15 leucocitos por campo, resto normal.
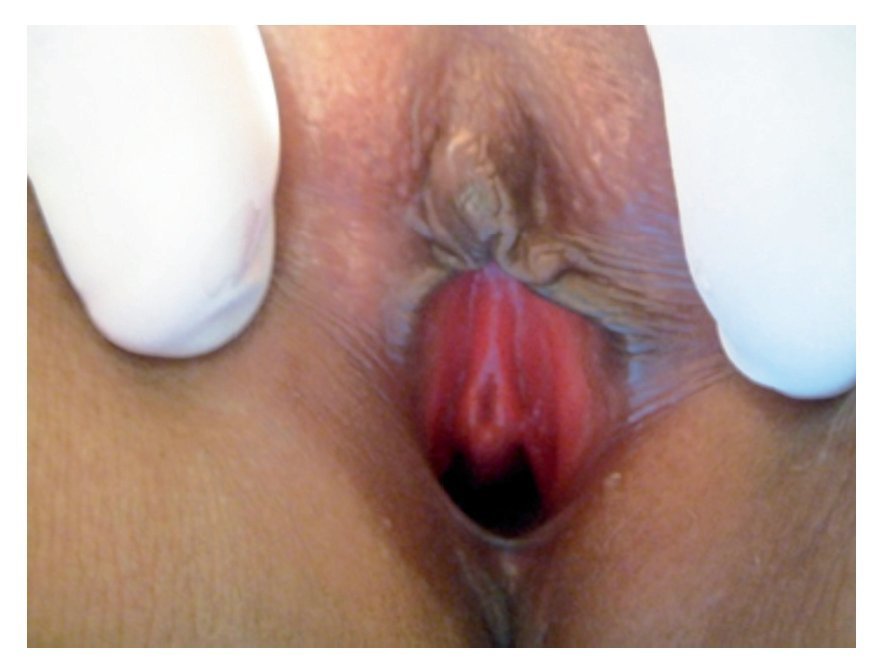
Imagen 1. Genitales externos con ausencia de vello púbico, clítoris, canal vaginal y meato uretral en vestíbulo.
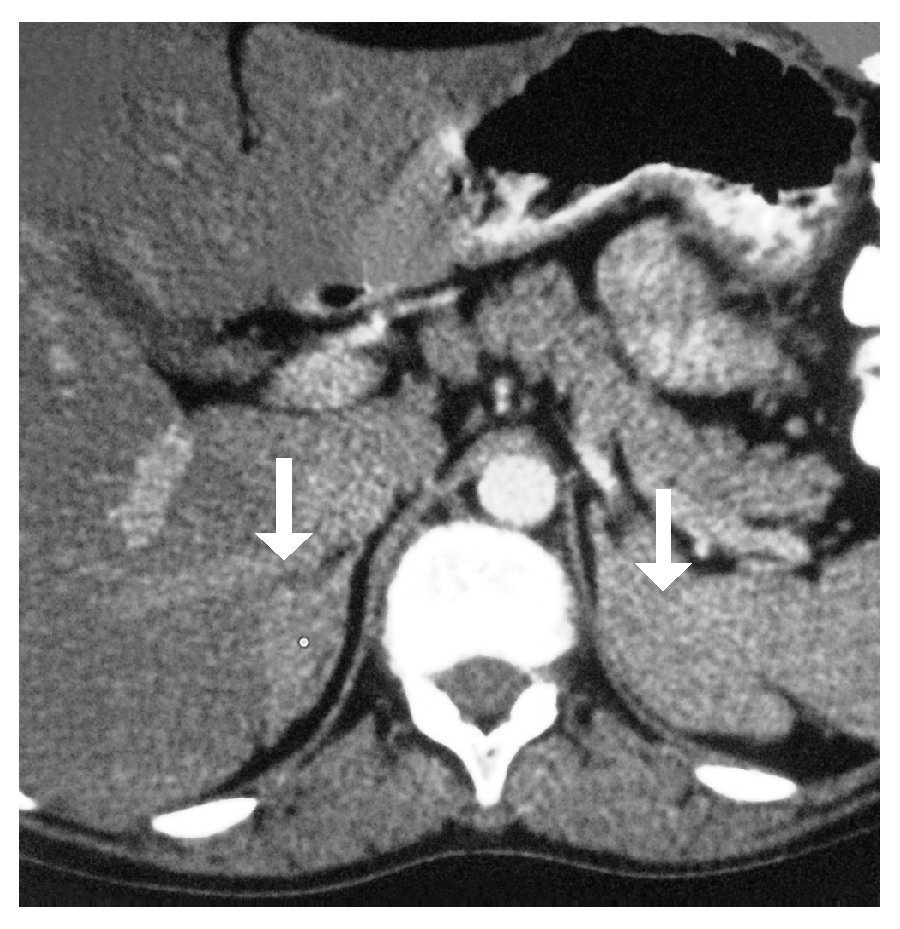
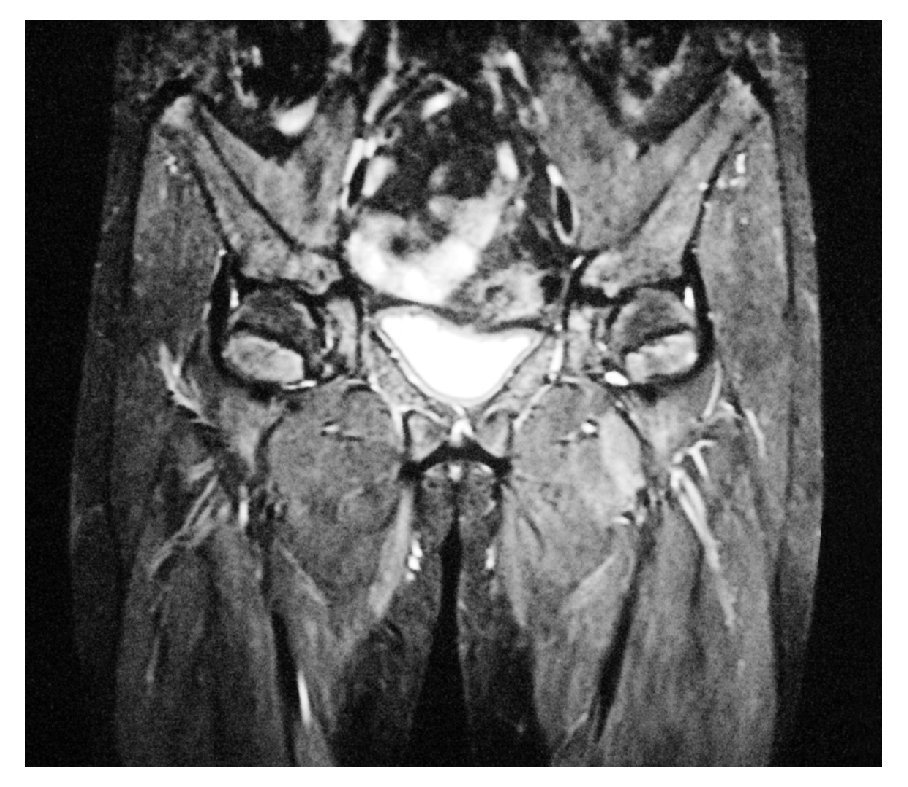
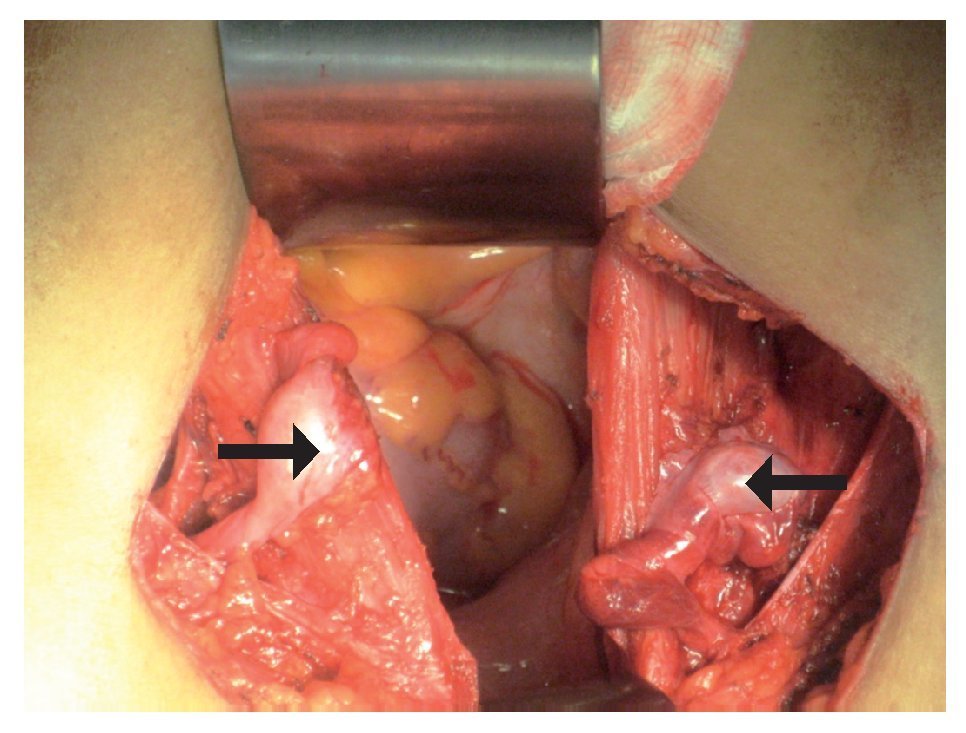
Electrocardiograma: ritmo sinusal, frecuencia cardiaca de 60 por minuto, eje con desviación a la izquierda, onda T invertida en derivaciones precordiales. Perfil hormonal con LH: 41.1, FSH: 67.9, Prolactina: 24.4, Estradiol: 10, ACTH: 53, Progesterona: 4.1, Testosterona: 10.0, hormonas tiroideas normales. Resonancia magnética con presencia de disgenesia gonadal, hiperplasia suprarrenal bilateral, probable feminización testicular, ausencia de útero y ovarios, con alteración metabólica en médula de huesos largos (Imágenes 2 y 3). Se le realizó valoración por el servicio de genética, quienes sugirieron realizar tipificación de PCR para cromosoma Y, encontrando a paciente genotipicamente XY. Se complementó con diagnósticos sindromáticos de hipogonadismo, hiperaldosteronismo, hipokalemia e hipertensión arterial; se diagnosticó como deficiencia de la 17 a-hidroxilasa. Se le realizó orquiectomía de remantes testículares bilaterales, los cuales se localizaron preperitoneales, en orificio inguinal profundo de manera bilateral de tamaño de 1 cm por 1 cm (Imágenes 4 y 5), así como tratamiento con nifedipino, enalapril, espironolactona y sales de potasio. Finalmente se le aplicaron implantes mamarios.
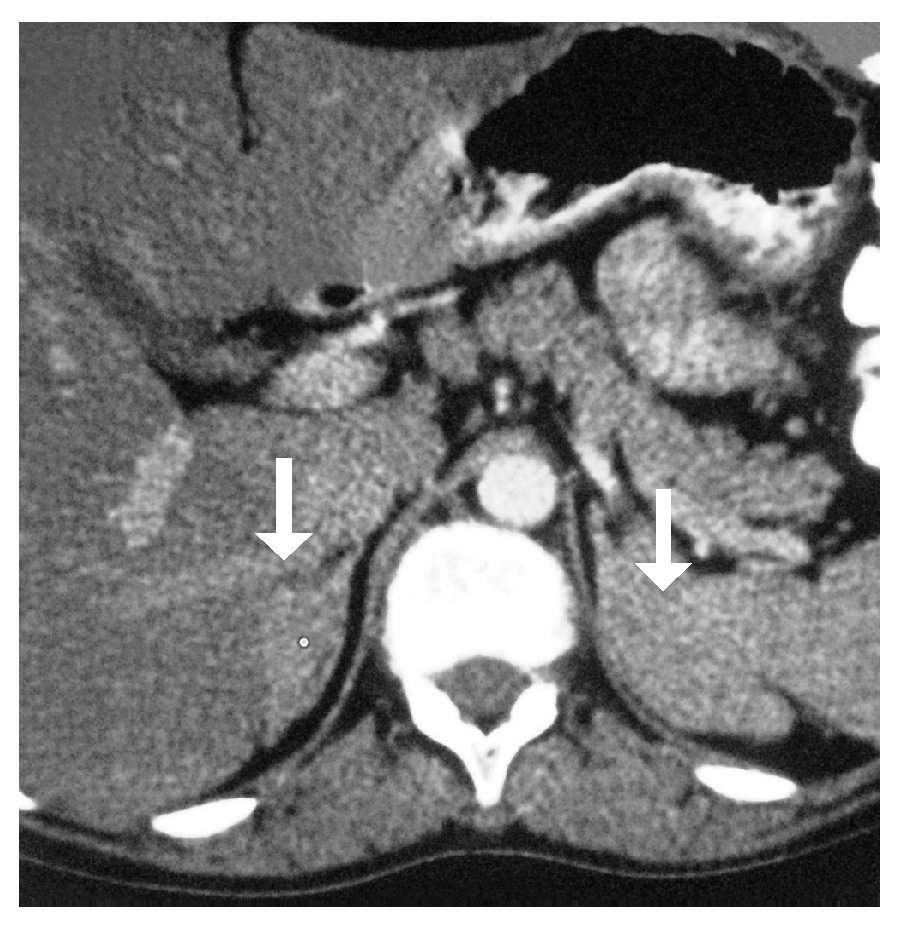
Imagen 2. Resonancia magnética nuclear, en la que se muestra hiperplasia suprarrenal bilateral.
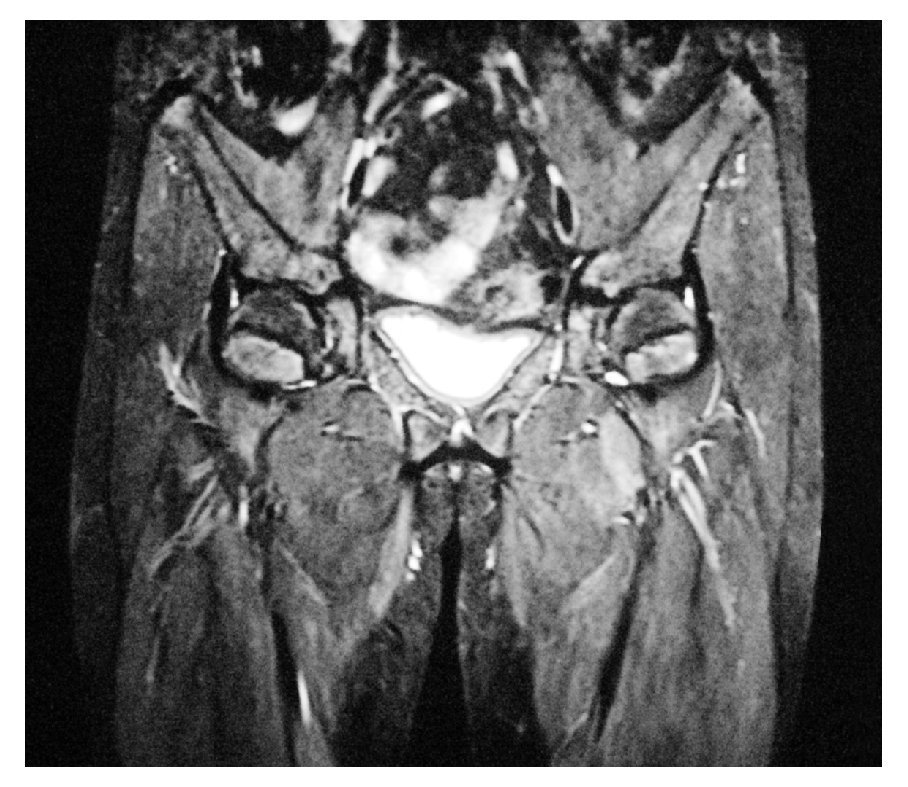
Imagen 3. Resonancia magnética nuclear, se evidencia la ausencia de útero y ovarios.
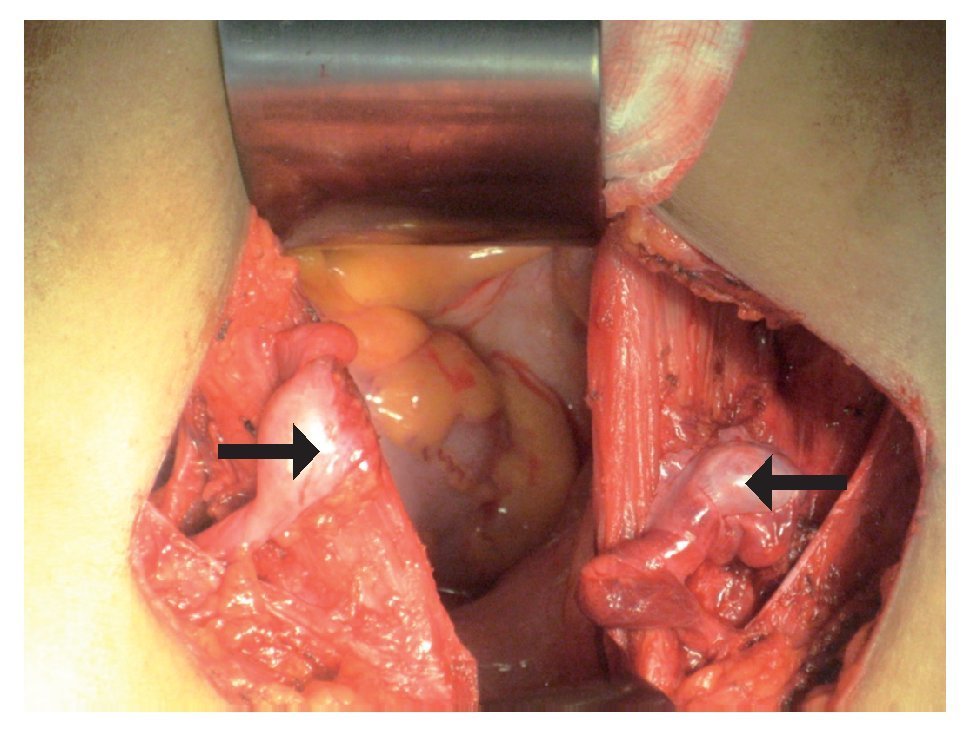
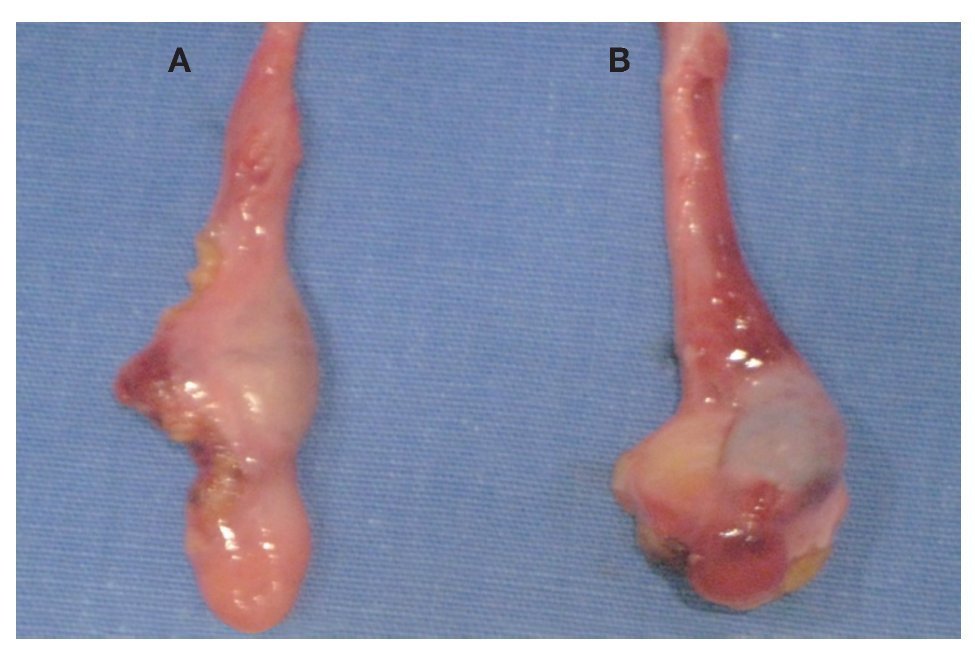
Imagen 4. Remantes testiculares bilaterales, los cuales se encontraban pre-peritoneales en orificio inguinal profundo de manera bilateral, con dimensiones de 1 cm por 1 cm.
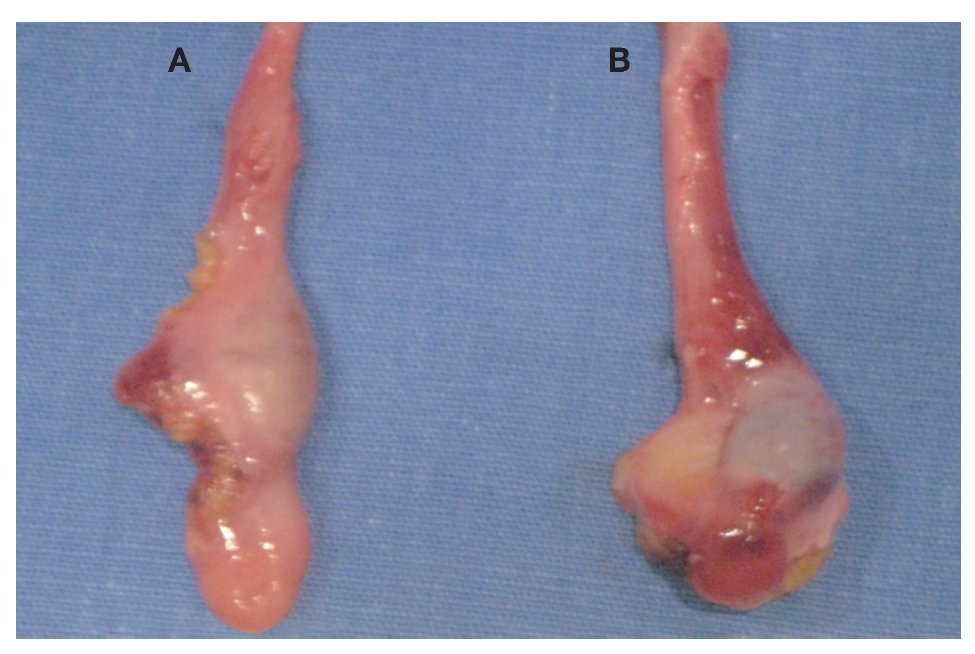
Imagen 5: Remantes testiculares. A. Gónada derecha. B. Gónada izquierda.
¿ PRESENTACIÓN DE CASO 2
Mujer de 15 años de edad, sin antecedentes de importancia. Amenorrea primaria, G-0. Negó inicio de actividad sexual. Inició su padecimiento actual al nacimiento. Su mamá notó ambigüedad de los genitales sin darle importancia, refirió crecimiento y desarrollo aparentemente sin alteraciones hasta que presentó bronconeumonía dos años antes valorada e interrogada en hospital de segundo nivel y posteriormente referida a nuestro hospital.
A la exploración física presentó signos vitales en límites normales, fenotípicamente con edad biológica mayor a la cronológica, sin fascies característica, con acné importante en cara, con nariz grande, cuello sin alteraciones; tórax anterior sin crecimiento mamario y el posterior normal; abdomen sin alteraciones. Genitales con distribución androide de vello púbico, se identificaron ambos testículos a nivel de los anillos inguinales superficiales, de aproximadamente 4 cm por 3 cm por 2 cm, ambos con epidídimos bien delimitables y conducto deferente identificable; se encontró falo de aproximadamente 3.5 cm por 1 cm, por 1 cm, identificándose cuerpos cavernosos; no se ubicó el meato urinario, los labios mayores identificables, sin labios menores; lo que correspondería al introito se canalizó, observándose salida de orina, resto de la exploración sin alteraciones. Exámenes de laboratorio en parámetros normales. USG: con ausencia de útero y ovarios, corroborándose testículos en canal inguinal. Testosterona: 2.16 ng/mL (H:0.2-0.8 M:3.7-9.5); DHT: 66 ng/mL (H:Menor de 200 M:200-800); Androstendiona: 6878.53 pg/mL (700-2000). Se valoró por genética, quienes evidenciaron cariotipo 46 XY y defecto enzimático en 17 b-hidroxiesteroide deshidrogenasa tipo 3. Se realizó orquiectomía bilateral.
¿ DISCUSIÓN
La hiperplasia suprarrenal congénita es un desorden resultado de la deficiencia de una de las cinco enzimas requeridas para la síntesis del cortisol en la corteza adrenal. La forma más común de la enfermedad es la clásica, debida a la deficiencia de la 21 hidroxilasa, seguida por la deficiencia de la 11 hidroxilasa, y más rara por deficiencia de la 17a-hidroxilasa.12-14
La disgenesia gonadal es una forma rara de hiperplasia suprarrenal congénita, con defecto en la biosíntesis del cortisol y esteroides sexuales que resultan en exceso de mineralocorticoides, hipertensión hipokalémica y anormalidades sexuales, presentándose en personas genéticamente hombres, con fenotipo femenino completo; menos frecuentemente, con genitales externos ambiguos, que se correlacionan con la actividad enzimática y la severidad del bloqueo de la 17 a-hidroxilasa y el subsecuente defecto en la síntesis de la testosterona.5,8,9,14 Históricamente se describió por primera vez en 1966 por Biglieri y colaboradores.5,6,9 Existe una mayor prevalencia en ciertos grupos étnicos, como los menonitas canadienses y los brasileños.4,7 Fisiológicamente, la esteroidogénesis suprarrenal es un proceso complejo y secuencial que involucra a una serie de enzimas que, actuando sobre el colesterol, producen una gran variedad de esteroides esenciales para la vida. El primer paso en la biosíntesis de todas las hormonas suprarrenales, es la conversión del colesterol a pregnenolona por acción del citocromo P450scc.4 Esta enzima es el punto de partida para la síntesis de glucocorticoides y mineralocorticoides. Su función es hidroxilar en posición 17 a la pregnenolona y a la progesterona, a la vez que actúa como liasa, rompiendo el enlace entre los carbonos 17 y 20 y con ello, permitiendo la síntesis de andrógenos sexuales.1,6,7
La hiperplasia suprarrenal congénita tiene un patrón autosómico recesivo, causado por mutaciones en el gen de la citocromo P450c17(CYP17) que media la actividad de la 17 a-hidroxilasa y la 17,20 liasa.3,6 El análisis de la actividad enzimática muestra que es necesario más de 25% de actividad normal para una masculinización normal de los genitales externos. En la deficiencia de la 17 a-hidroxilasa, la disminución de la secreción de cortisol causa aumento de la producción de ACTH, lo que resulta en la sobreproducción de 17 desoxiesteroides por la corteza adrenal. La progesterona sérica es un marcador en el diagnóstico.5,7 Después de la estimulación con ACTH, el nivel de progesterona se altera. Muchos pacientes tienen una baja o anormal producción de aldosterona.7 El diagnóstico suele hacerse en el momento de la pubertad, por la presencia de hipertensión arterial (aunque 10% a 15% de los pacientes pueden ser normotensos), hipokalemia (con síntomas neuromusculares e intestinales) e hipogonadismo hipergonadotrófico (amenorrea primaria y ausencia de caracteres sexuales secundarios en la mujer y pseudohermafroditismo masculino), usualmente no hay síntomas de insuficiencia adrenal.1,6 Nuestro primer paciente se presentó con el cuadro clínico característico, sin embargo el segundo no presentó sintomatología alguna. Los casos femeninos, poseen genitales externos femeninos al nacimiento, pero sin desarrollo sexual durante la pubertad; los casos masculinos tienen genitales externos femeninos, canal vaginal ciego y ausencia de útero y trompas de Falopio.9 Como lo observado en nuestros casos. El diagnóstico se puede establecer basado en los niveles elevados de 17-deoxi-C21 esteroides, progesterona, pregnenolona, DOC y corticosterona séricas y un incremento en la excreción urinaria de sus metabolitos por medio de RIA, cromatografía/espectroscopia.9
Por lo general la hipertensión se resuelve con glucocorticoides, pero persiste si el diagnóstico es erróneo. Los antagonistas de mineralocorticoides, como la espironolactona, pueden ayudar a un mejor control de la tensión arterial. El añadir un bloqueador de los canales del calcio es efectivo, si la hipertensión persiste.5 En nuestra primera paciente la hipertensión y la hipokalemia se resolvieron después del reemplazo con glucocorticoides y el tratamiento con diuréticos ahorradores de potasio; en estos casos se deben adicionar esteroides sexuales para compensar el déficit secundario a la baja producción gonadal.15 En nuestros casos se utilizó el reemplazo con hormonas sexuales, para inducción del desarrollo sexual. La cirugía genital es una de las intervenciones más controvertidas en el manejo de intersexo, se suele realizar una cirugía correctiva para resecar o crear los órganos reproductores apropiados para el sexo del niño.14,16,17 Se recomienda una gonadectomía bilateral tan pronto como sea posible, ya que la gónada disgenésica tiene alto potencial de malignización (aproximadamente un tercio de los pacientes desarrolla un gonadoblastoma durante la primera a cuarta década de la vida).16,17 Por tal motivo a nuestra primera paciente se le realizó gonadectomía profiláctica. La correcta determinación del sexo es importante no sólo en lo que respecta al tratamiento, sino también en lo relacionado con el bienestar emocional del niño.16
¿ CONCLUSIÓN
Los recién nacidos con anomalías del desarrollo de los genitales externos deben ser diagnosticados tempranamente para minimizar las complicaciones medicas, psicológicas y sociales. La deficiencia de 17 a-hidroxilasa es una rara condición, debe ser sospechada en pacientes con hipertensión hipokalémica y retardo en el desarrollo de los caracteres sexuales secundarios, para lo que debe implementarse una terapia apropiada. En estas pacientes se les asignó el sexo femenino, por lo que se les manejó con reemplazo hormonal con base en estrógenos, para la inducción de los caracteres sexuales secundarios. La gonadectomía profiláctica está indicada en hombres genéticos con deficiencia de la 17 a-hidroxilasa por el riesgo de malignizarse. Y pueden localizarse intraabdominales, en el canal inguinal o labio-escrotales.
Correspondencia: Dr. Jesús Emmanuel Rosas Nava.
Insurgentes Sur 4411, Residencial Insurgentes Sur, Tlalgoligia, Tlalpan, D. F. 14430.
Teléfono: 5485 6435.
Coreo electrónico:rosas_nava_jes@hotmail.com