


El síndrome de abdomen en ciruela pasa es una rara malformación que se presenta en uno de 40,000 nacidos vivos. Casi exclusivamente en varones (>95%), y se caracteriza por la triada que incluye: anomalías del tracto urinario, deficiencia de la musculatura abdominal y criptorquidia bilateral. La pared abdominal protruyente semeja una ciruela seca.
Presentamos el caso de un paciente varón de 2 años 4 meses. El abdomen era prominente, con dibujo de asas intestinales, deficiencia de la musculatura central abdominal y testículos no descendidos. Presentaba reflujo vesicoureteral bilateral grado v, vejiga de gran capacidad y mega-uréteres. Realizamos reconstrucción de la pared abdominal tipo Monfort, orquidopexia bilateral y remodelación con reimplante ureteral bilateral tipo Cohen. En la evolución posquirúrgica observamos una reducción de la circunferencia abdominal, disminución de la ectasia urétero-piélica, corrección del reflujo ureteral y permanencia de los testículos en escroto.
Un tercio de los pacientes progresarán a falla renal. El grado de displasia, la enfermedad quística y una creatinina sérica basal>0.7mg/dl son factores de riesgo para el deterioro renal. La reconstrucción de la pared abdominal tiene un fin estético; se ha observado también mejoría en el vaciamiento vesical, intestinal y en la función pulmonar. Se recomienda la orquidopexia temprana.
Prune belly syndrome is a rare malformation that presents in 1 out of every 40,000 live births. It occurs almost exclusively in males (>95%) and is characterized by the following triad: urinary tract anomalies, abdominal muscle deficiency, and bilateral cryptorchidism. The protruding abdominal wall resembles a dried prune.
A 28-month-old boy presented with a prominent abdomen, visible outline of the intestinal segments, central abdominal muscle deficiency, and undescended testes. He had grade V bilateral vesicoureteral reflux, a large-capacity bladder, and megaureters. We performed abdominal wall reconstruction with the Monfort technique, bilateral orchidopexy, and ureteral remodeling with a bilateral Cohen ureteral reimplantation. Reduced abdominal circumference, reduced ureteropelvic ectasia, corrected ureteral reflux, and retention of the testes in the scrotum were observed in the postoperative progression.
One-third of these patients progress to renal failure. The grade of dysplasia, cystic disease, and baseline serum creatinine>.7mg/dl are risk factors for renal deterioration. Abdominal wall reconstruction is performed for esthetic purposes, but improved bladder and bowel emptying and pulmonary function have also been suggested to result. Early orchidopexy is recommended.
El síndrome de abdomen en ciruela pasa (SACP) es una rara malformación que se presenta en uno de 40,000 nacidos vivos, casi exclusivamente en varones (>95%), y se caracteriza por la triada que incluye: anomalías del tracto urinario, deficiencia de la musculatura abdominal y criptorquidia bilateral. La pared abdominal protruyente e hipoplásica semeja una ciruela seca, de ahí el nombre del síndrome. Las malformaciones del tracto urinario son debidas a la displasia del músculo liso presente en la pelvis y los uréteres. Dependiendo del tipo y de la severidad tiene 3 diferentes formas clínicas de presentación: 1) la forma oligúrica no viable con displasia renal severa; 2) displasia renal marcada, mega-uréteres, mega-vejiga, falla renal progresiva; y 3) la forma más favorable, con displasia renal moderada y distintos grados de dilatación ureteral y vesical1–3.
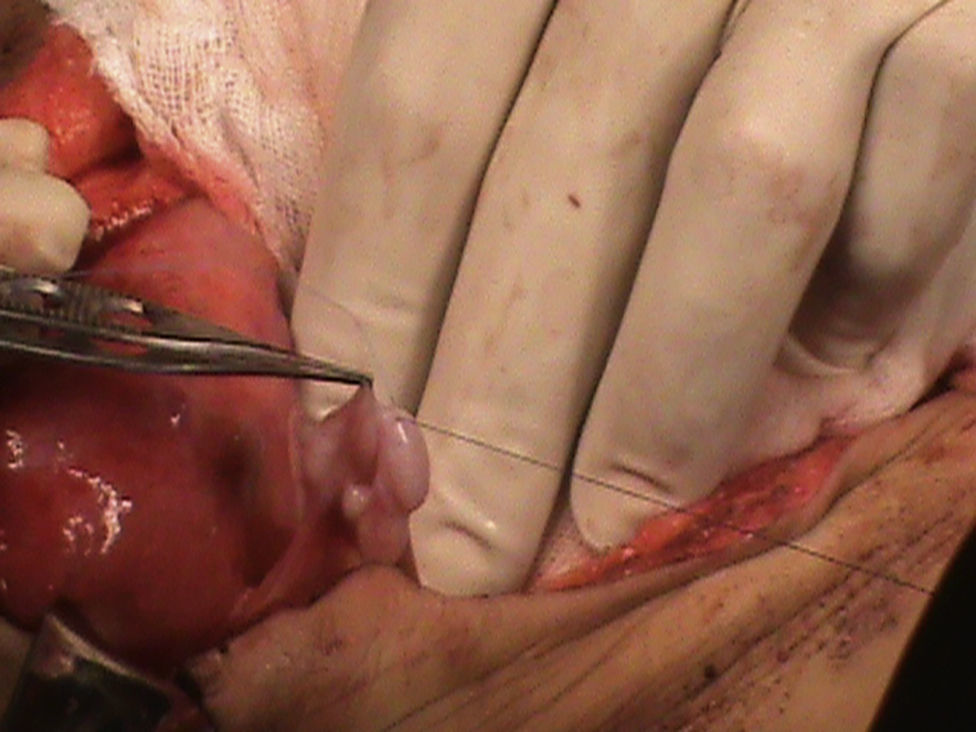
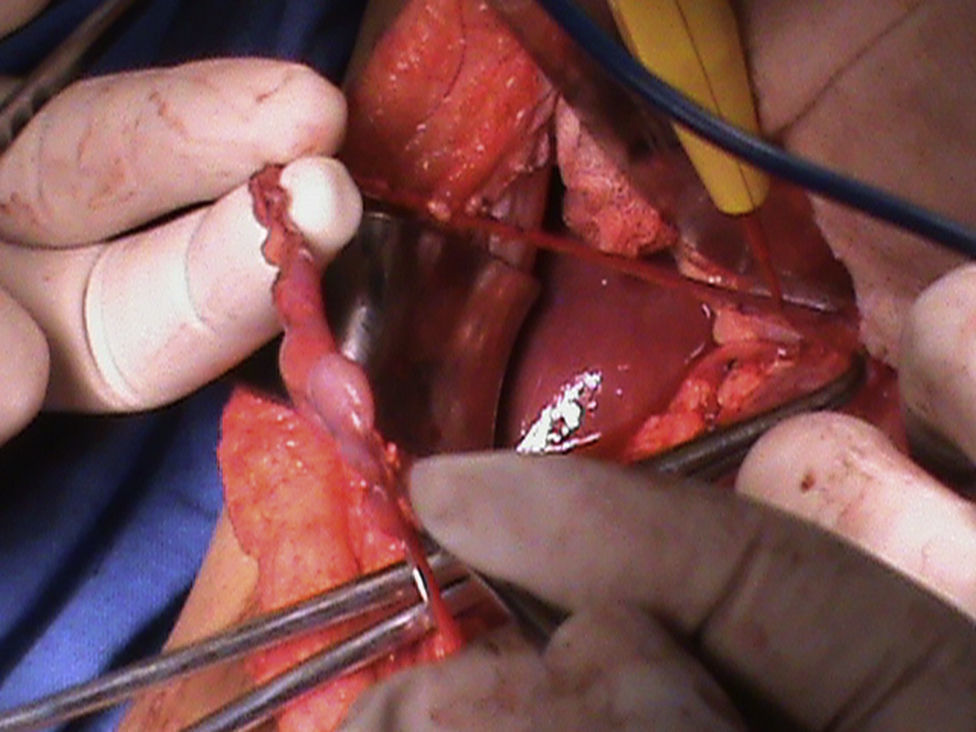
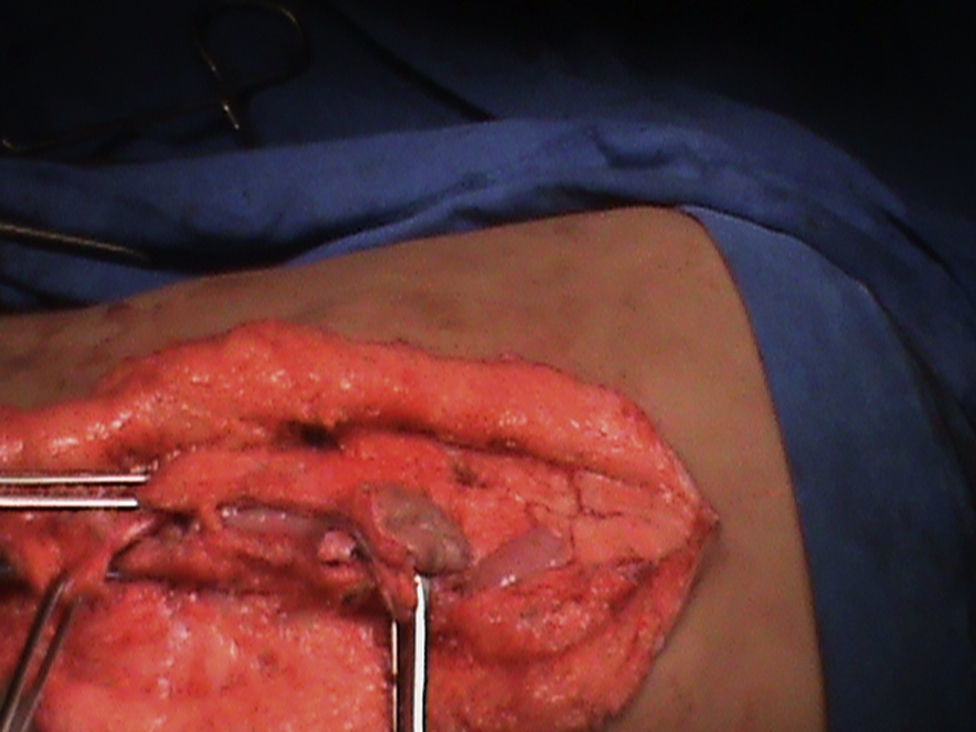
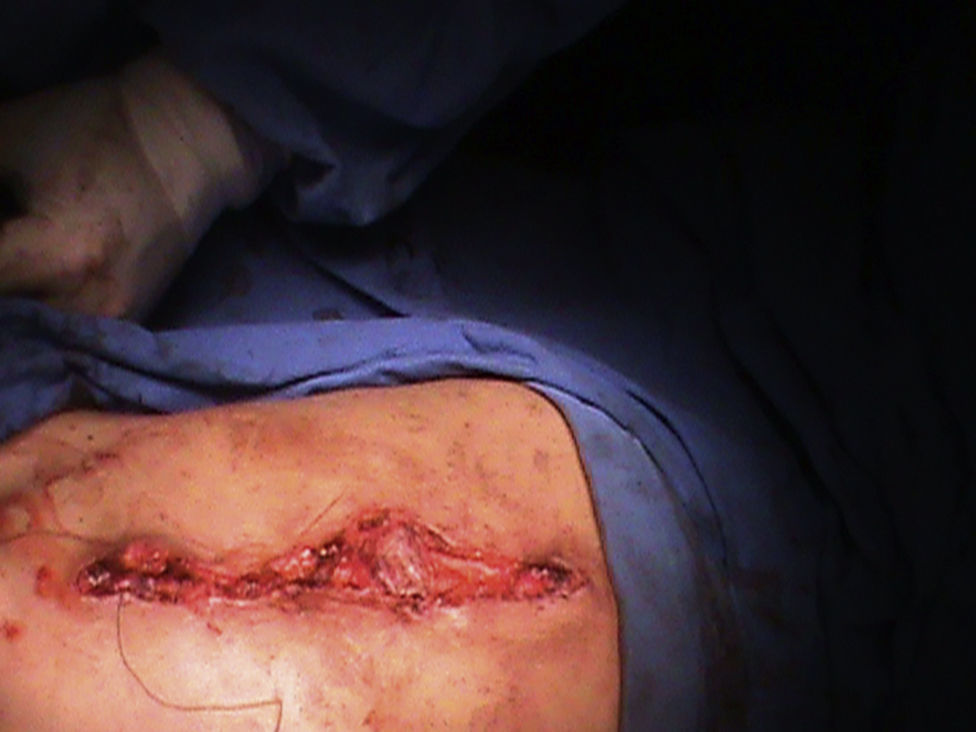
Presentación del casoPresentamos el caso de un paciente varón de 2 años 4 meses, el cual es diagnosticado en el segundo trimestre de la gestación de ectasia renal bilateral; obtenido vía cesárea, se le realizó vesicostomía al mes de edad, presenta displasia bilateral de cadera. A la exploración física con abdomen globoso, abultado en los flancos, prominente, dibujo de asas intestinales sobre la piel, se palpa deficiencia de la musculatura en la línea media de la pared abdominal, y no se palpan testículos en el escroto. Presencia de reflujo vesicoureteral bilateral grado v, megavejiga y megauretéres en el cistograma, así como hidronefrosis bilateral. Al llegar a nuestro servicio presenta un nivel de creatinina de 1.9mg/dl, urea de 143.4mg/dl, depuración de creatinina de 17.6ml/min/1.73 por fórmula de Schwartz, insuficiencia renal grado iv. Al paciente se le localizó los testículos intra-abdominales y se efectuó orquidopexia bilateral en 1 etapa (figs. 1 y 2), posteriormente se realizó reconstrucción del abdomen con la técnica de Monfort (figs. 3 y 4) y la remodelación ureteral con reimplante bilateral tipo Cohen. Cursa con adecuada evolución posquirúrgica, con una reducción de la circunferencia abdominal, disminución de la ectasia urétero-piélica, corrección del reflujo ureteral bilateral, adecuado vaciamiento vesical y permanencia de los testículos en el escroto.




Recientemente se sugirió la implicación del gen hepatocyte nuclear factor 1 beta en la etiología del síndrome, basándose en 2 casos reportados. Granberg et al. descartaron la posibilidad de la implicación de este gen al detectarlo solamente en uno de 32 pacientes estudiados, por lo cual sigue sin determinarse la base genética de esta enfermedad4.
Cerca de un tercio de los pacientes que sobreviven al periodo posnatal progresarán a falla renal, necesitando un trasplante renal en el futuro. Los factores de riesgo identificables tempranamente son el grado de displasia renal y la enfermedad quística, siendo el marcador más especifico de displasia significativa la creatinina basal, que es el nivel más bajo de creatinina en los primeros 12 meses de vida. Noh et al. reportaron que la creatinina basal>0.7mg/dl, así como la pielonefritis clínica, son factores pronósticos de falla renal5,6. En el caso de nuestro paciente ya presentaba una creatinina sérica de 1.9mg/dl, que se traduce como mal prónostico de la función renal.
Las malformaciones ortopédicas oscilan entre 30-40% de los pacientes, y el sistema osteomuscular es el segundo afectado en frecuencia después del genitourinario en este síndrome, la displasia de cadera y la atrofia de alguna extremidad son comunes1.
La apariencia del abdomen es debida a la displasia e hipoplasia de los músculos abdominales centrales, se ha observado por microscopia electrónica un desorden de los miofilamentos y desorganización de la línea Z, las 3 capas musculares son afectadas, particularmente debajo del ombligo, donde el músculo puede ser reemplazado completamente por tejido fibroso, en contraste los músculos periféricos están parcial o totalmente respetados5.
La base de la reconstrucción de la pared abdominal es el avance de la musculatura periférica bien inervada y vascularizada, con o sin la escisión de los músculos y fascia afectados5. La reconstrucción de la pared abdominal tiene un fin estético, también se ha descrito mejoría en el vaciamiento vesical, intestinal y en la función pulmonar debido a la mayor presión intraabdominal que puede ser ejercida con la reconstrucción, sin embargo hacen falta estudios para fundamentar mejor estos últimos beneficios. Existen distintas técnicas para realizar la reconstrucción de la pared abdominal; en la técnica de Monfort se realiza una incisión elíptica desde el apéndice xifoides hasta la sínfisis del pubis, una segunda incisión se realiza en el ombligo para que permanezca in situ, se extiende la disección de la fascia y el músculo hasta la línea axilar anterior, se realizan 2 incisiones verticales sobre la fascia a los lados de las arterias epigástricas superiores, dejando un puente central de fascia, luego se avanza la fascia de ambos lados sobre el puente central aumentando el grosor de la pared abdominal y reduciendo el tejido redundante. La técnica fue descrita en 1991 y efectuada en 9 pacientes con resultados satisfactorios en todos ellos7–9.
La reconstrucción ureteral está indicada en la presencia de un deterioro progresivo de la función renal o persistencia de infecciones en las vías urinarias, con el fin de reducir la estasis urinaria1.
El sistema colector renal está característicamente dilatado, pero el grado de dilatación no se relaciona con el grado de displasia. El reflujo vesicoureteral está presente en el 75% de los niños con SACP9.
La criptorquidia bilateral con testículos abdominales es una de las principales características del SACP, la obstrucción mecánica producida por la dilatación de la vejiga y el sistema urinario, sumado a la deficiencia en la pared abdominal, parecen ser los factores clave en la falta del descenso testicular. El gubernáculo, igualmente importante en el descenso, se ha notado atrésico o anormalmente adherido al tubérculo púbico9.
Se recomienda la orquidopexia temprana en estos pacientes, alrededor de los 6 meses, en el momento de realización de las demás cirugías de corrección que fueran necesarias1,9.
ConclusionesContinúa sin determinarse la base genética del SACP. Es común la evolución hasta la falla renal en estos pacientes, la presencia de una creatinina sérica>0.7mg/dl es un factor pronóstico importante de la función renal. Las anormalidades ortopédicas son las segundas en frecuencia después de las del sistema genitourinario y la pared abdominal. Hasta ahora la reconstrucción de la pared abdominal tiene un fin estético, y se sugieren beneficios en el vaciamiento vesical, intestinal y en la función pulmonar que deberán ser comprobados con más estudios; la técnica de Monfort es una buena opción para la reconstrucción. La reconstrucción ureteral está indicada en la presencia de un deterioro progresivo de la función renal o persistencia de infecciones en las vías urinarias, con el fin de reducir la estasis urinaria. Se recomienda la orquidopexia temprana en estos pacientes, alrededor de los 6 meses, en el momento de realización de las demás cirugías de corrección.
Responsabilidades éticasProtección de personas y animalesLos autores declaran que para esta investigación no se han realizado experimentos en seres humanos ni en animales.
Confidencialidad de los datosLos autores declaran que han seguido los protocolos de su centro de trabajo sobre la publicación de datos de pacientes.
Derecho a la privacidad y consentimiento informadoLos autores han obtenido el consentimiento informado de los pacientes y/o sujetos referidos en el artículo. Este documento obra en poder del autor de correspondencia.
FinanciaciónLos autores no recibieron ningún patrocinio para llevar a cabo este estudio.
Sin fuente de financiación.
Conflicto de interesesLos autores declaran no tener conflicto de intereses.
A mi esposa, por su comprensión y apoyo incondicional. A mis padres y hermanos por creer siempre en mí. A mis maestros por su dedicación a mi formación. Al Dr. Julio Gómez por sus invaluables enseñanzas en el campo de la urología pediátrica.