


Introducción
La enfermedad de Castleman (EC) es un desorden linfoproliferativo poco frecuente. Benjamín Castleman la describió por primera vez en 19561,2. Puede aparecer en una gran variedad de localizaciones, asociada a una sobreestimulación antigénica de causa desconocida1.
Clínicamente se clasifica como: localizada y diseminada1,3. Presenta dos variedades histológicas: una hialinovascular (91%) y otra plasmocelular1,4,5. El diagnóstico diferencial debe realizarse con enfermedades que se presenten con grandes masas mediastínicas1. El tratamiento quirúrgico es diagnóstico y a la vez curativo en las formas localizadas, mientras que son necesarios tratamientos complementarios en las formas multicéntricas6,7.
Se describen dos casos de EC localizadas en el tórax, en el Servicio de Cirugía Torácica del Hospital Universitario La Fe de Valencia en los últimos 10 años, y se realiza una revisión de la bibliografía.
Paciente 1
Mujer de 33 años, sin alergias conocidas, fumadora de un paquete de cigarrillos al día, intervenida a los 14 años de apendicectomía y a los 21 años, de escoliosis. Presentó cuadro clínico de dolor retroesternal opresivo, irradiado a espalda, acompañado de disnea y febrícula de varios días de evolución. Acudió a su hospital de referencia donde se ingresó para estudio y tratamiento, presentando una mejoría de la sintomatología con antiinflamatorios no esteroideos.
En el examen físico, se encontró consciente, orientada, bien hidratada, con eritema malar bilateral y telangiectasias en cara y cuello. Sin adenopatías periféricas. En la auscultación cardíaca con tonos rítmicos, sin soplo. En la auscul tación respiratoria se observó murmullo vesicular sin ruidos añadidos. Abdomen blando, depresible, doloroso a la palpación en hipocondrio derecho, con hepatomegalia de 2-3 cm. Sin otro hallazgo a resaltar.
En el análisis de sangre realizado se observaron leucocitos de 10.600/l con fórmula normal, hemoglobina de 10,6 g/dl y hematocrito del 31%, una velocidad de sedimentación globular de 129 mm/h, y con perfil hepático y renal dentro de la normalidad. Dosificación de inmunoglobulinas (Ig): IgG 1.717 mg/dl, IgA 5.200, IgM 33. Inmunofijación en suero: intensa banda policlonal de IgA kappa y lambda, bandas policlonales IgG kappa y lambda y muy débil banda monoclonal IgG kappa. Inmunología: anticuerpo antinuclear positivo débil, C3 y C4 normal, anticuerpos anticardiolipina negativos. Serología: virus de la inmunodeficiencia humana negativo, citomegalovirus y virus de Epstein-Barr positivos para IgG y negativos para IgM. Hepatitis B y C negativos.
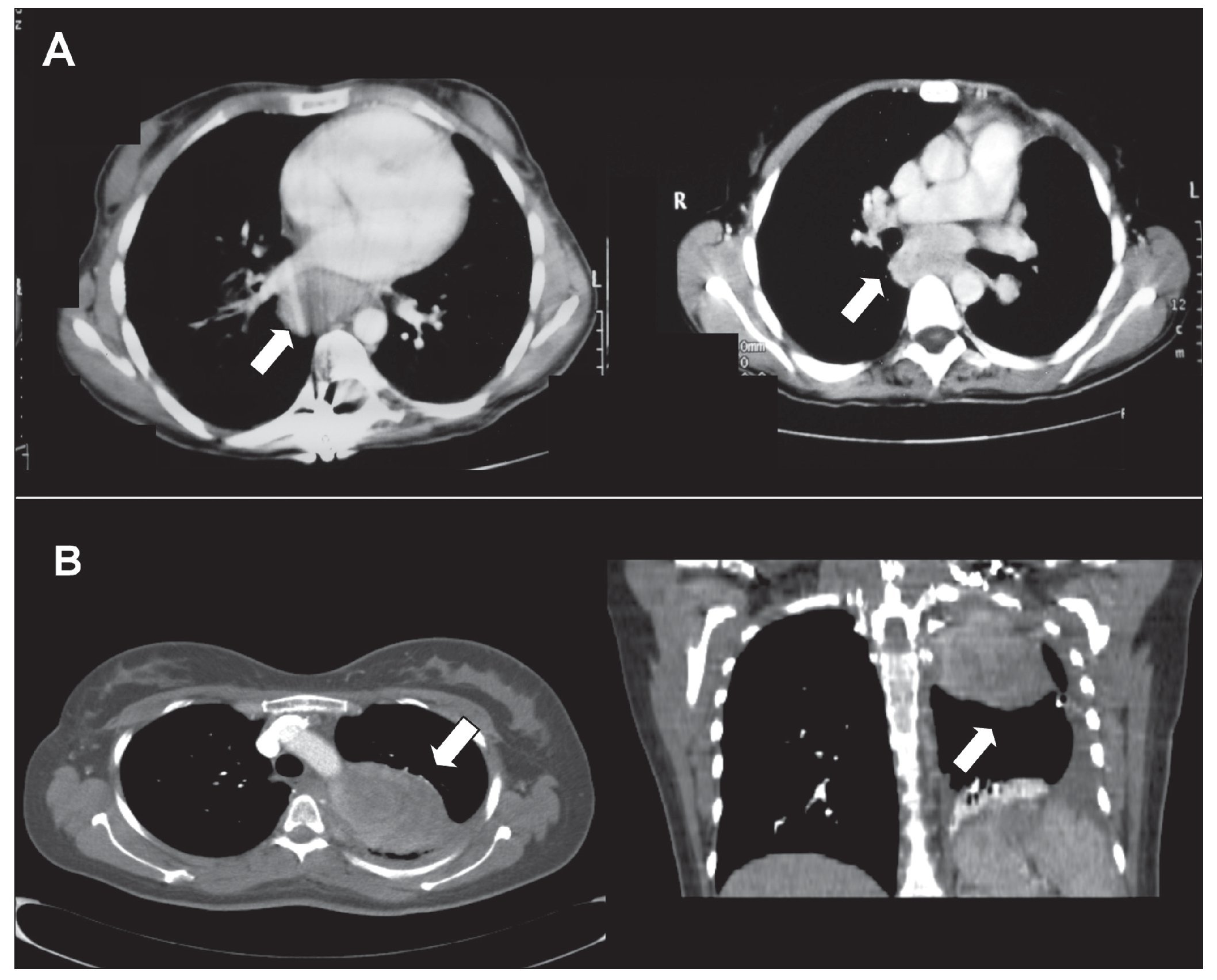
Entre las pruebas complementarias se describió: una radiografía de tórax con ensanchamiento mediastínico antero-posterior. En las radiografías seriadas óseas, no se observaron lesiones líticas. En la tomografía computarizada (TC) toracoabdominal (fig. 1A) se apreciaron adenopatías mediastínicas prevasculares y en espacio subcarinal, hepatomegalia homogénea sin lesiones focales y riñones aumentados de tamaño a predominio del lado derecho.
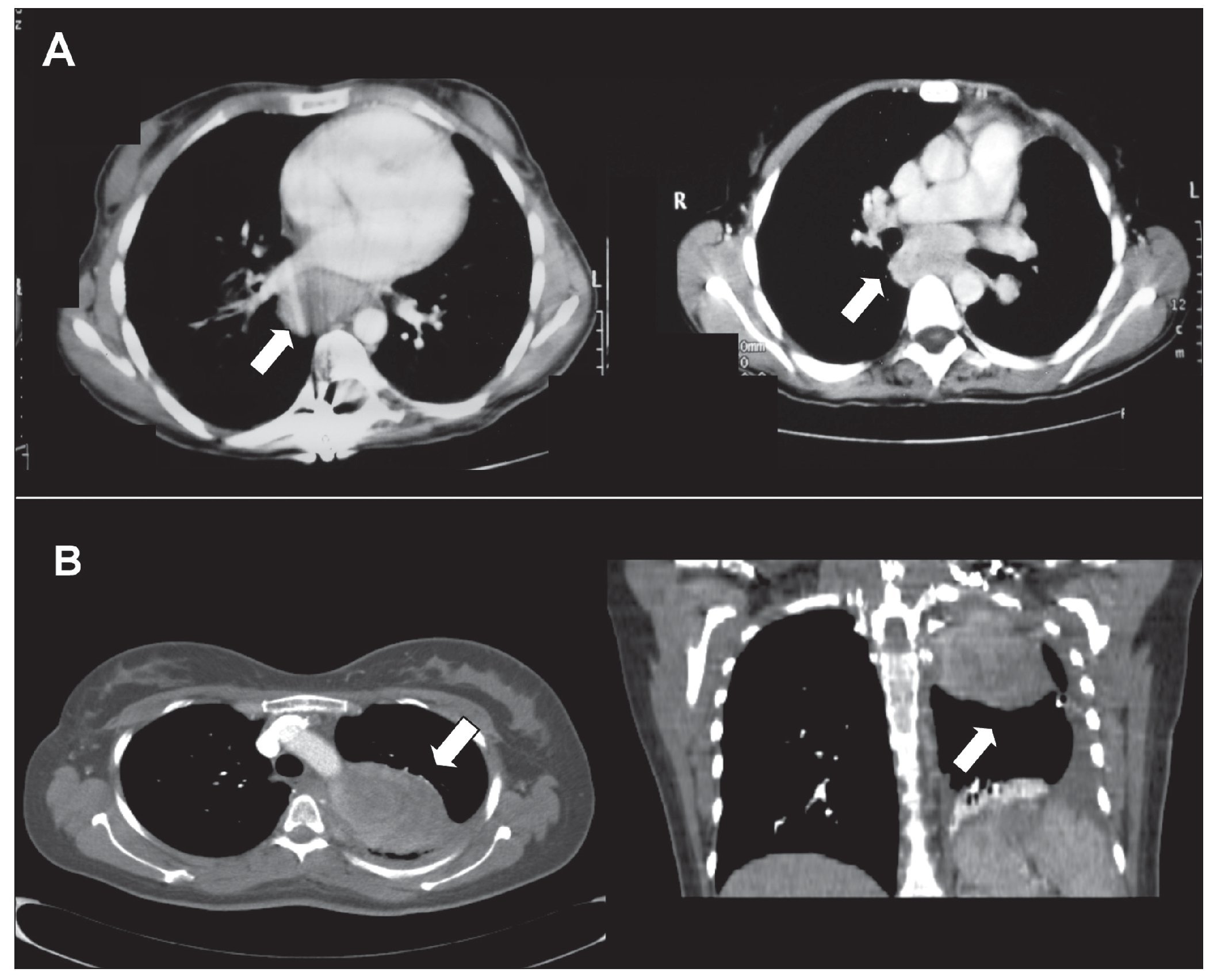
Figura 1.A. Tomografía computarizada. A la izquierda, adenopatía mediastínica prevascular de 4 ´ 5 cm; a la derecha, adenopatía en espacio subcarinal de 6 ´ 3 cm. B. Tomografía computarizada. Masa sólida heterogénea en hemitórax izquierdo de 8 ´ 6 ´ 7 cm con áreas de necrosis en su interior. A la izquierda, corte transversal, y a la derecha, corte coronal.
El aspirado de médula ósea mostró celularidad aumentada, de aspecto reactivo, con marcada eosinofilia y un 20-27% de células plasmáticas de aspecto maduro. La biopsia del ganglio mediastínico realizada por mediastinoscopia informó de linfadenitis crónica y plasmocitosis marcada.
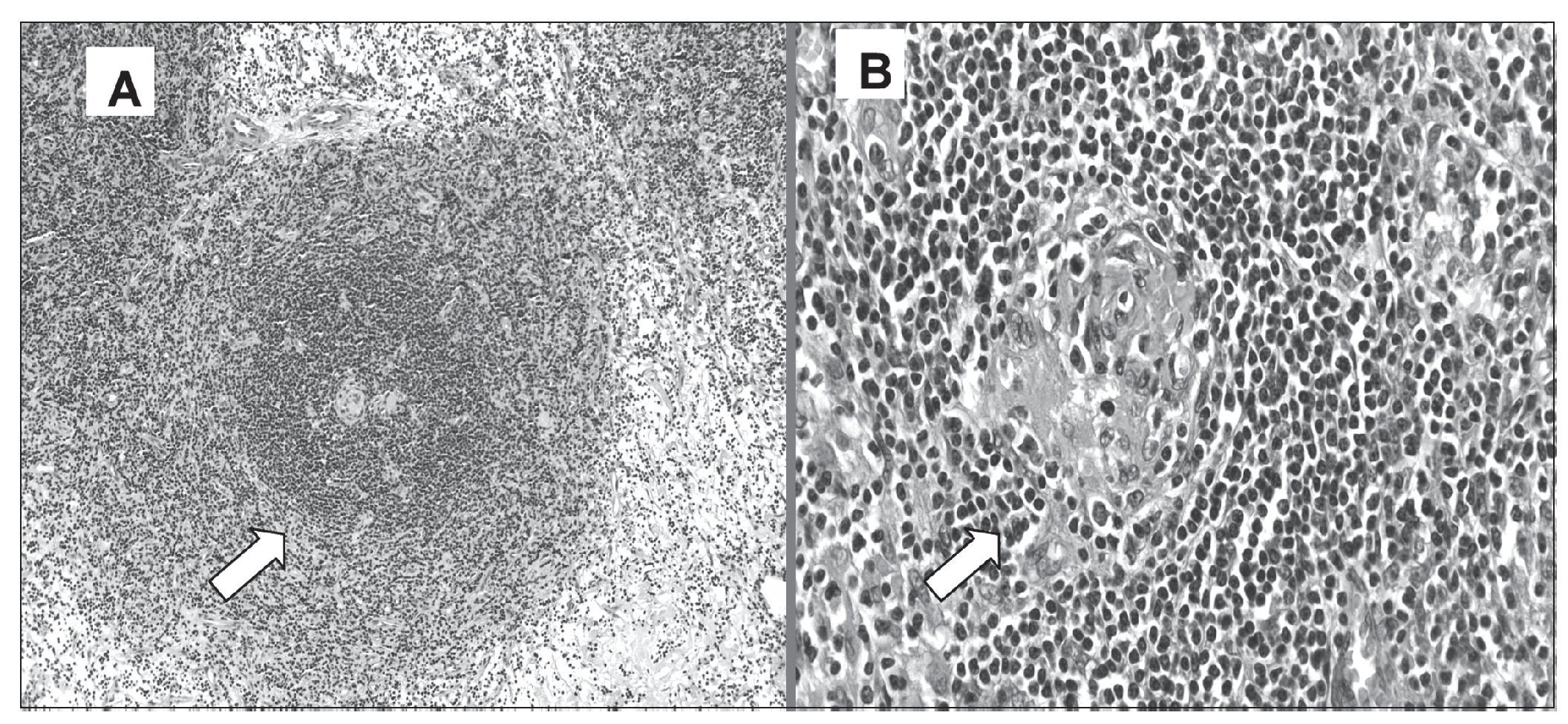
Con todos estos datos, sin diagnóstico claro de enfermedad hematológica, la paciente fue enviada a nuestro hospital para extirpación de la masa mediastínica por toracotomía izquierda, y se realizó una resección de masa ganglionar que llegaba a vértice con importantes ramas vasculares, precisando apertura de la pleura mediastínica retrocardíaca para conseguir la extirpación completa, que se extendía hasta el bronquio intermediario derecho. El examen de anatomía patológica de la pieza mostró EC variante plasmocelular (fig. 2A) cuyo fenotipo expresó policlonalidad (IgG+, kappa+, lambda +), en asociación de enfermedad de Hodgkin (CD15+, CD30+).
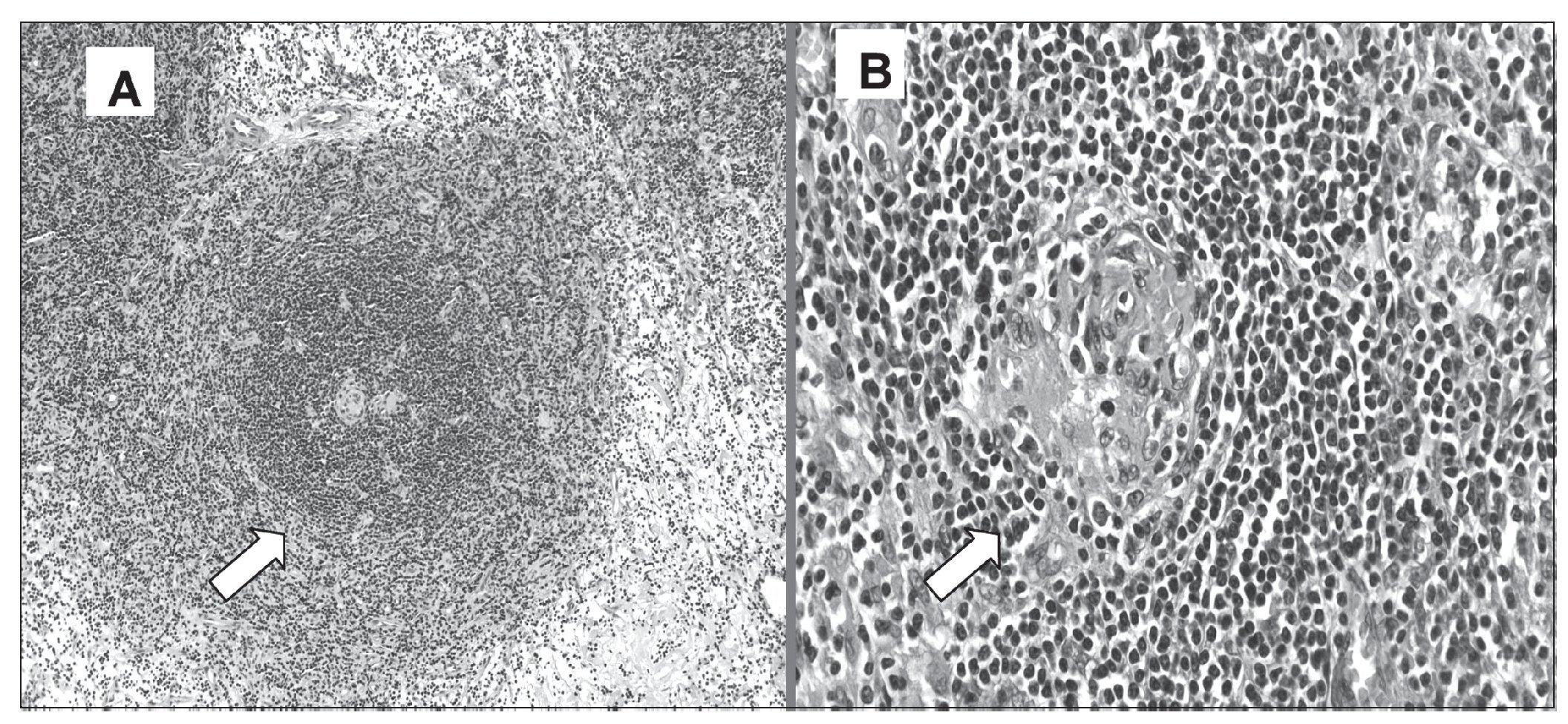
Figura 2.A. Histología de la pieza quirúrgica adenopatía mediastínica, que presenta folículos con zonas muy hiperplásicas, formada por anillos celulares concéntricos de linfocitos pequeños, células plasmáticas maduras y pequeños vasos sanguíneos. Hematoxilina - eosina x 40. B.- Histología: centros germinales hiperplásicos, con folículos hialinos vasculares que presentan vasos de paredes hialinas. Hematoxilina - eosina x 100.
Posteriormente, se trató este linfoma con quimioterapia y, actualmente, desde hace 8 años se encuentra libre de enfermedad.
Paciente 2Mujer de 32 años, alérgica a la penicilina, con antecedente de asma bronquial, que presentó dolor cervical asociado a disnea progresiva y tos seca de 15 días de evolución, por lo que acudió a Urgencias donde se realizó una radiografía de tórax en la que se detectó opacidad en hemitórax izquierdo con componente atelectásico y fue ingresada para completar el estudio. En la exploración física, se encontró a una paciente consciente, orientada, sin focalidad neurológica, con auscultación cardíaca de tonos rítmicos, sin soplo. En la auscultación respiratoria, se observó murmullo vesicular disminuido en hemitórax izquierdo. Abdomen blando, depresible, no doloroso, sin megalias ni masas.
Analíticamente todos los parámetros se encontraban dentro de la normalidad, incluido anticuerpo antinuclear negativo, factor reumatoide y antígeno carcinoembrionario normal. En la radiografía de tórax se observó opacidad en hemitórax izquierdo con derrame pleural. En la TC toracoabdominal pélvica se describió masa sólida heterogénea en lóbulo superior izquierdo de 8 ´ 6 ´ 7 cm, con atelectasia segmentaria en lóbulo inferior izquierdo, con derrame pleural, sin alteraciones en parénquima pulmonar o resto de estructuras mediastínicas; no se observaron alteraciones en el ámbito abdominal (fig. 1B). En la resonancia magnética (RM) se observó masa mediastínica posterior que impronta sobre ápex de hemitórax izquierdo heterogénea en la secuencia T2 y pequeñas zonas quístico-necróticas en su interior, de contorno bien definido.
Se realizó toracoscopia en la que se detectó una masa de 5 cm aproximadamente con otras lesiones pequeñas adheridas al ángulo costal posterior de superficie lisa nacarada, dura; se extrajeron 5 l de líquido pleural y se tomó biopsia, la cual se informó en un principio como focos compatibles con tumor mesenquimal de aspecto benigno. La citología del líquido pleural fue negativa para células malignas.
En vista de los hallazgos encontrados y la falta de diagnóstico, se realizó toracotomía izquierda y resección tumoral completa mediante segmentectomía en lóbulo superior izquierdo y extracción de esta masa.
En el estudio anatomopatológico de la pieza quirúrgica, se describió tejido linfoide de aspecto reactivo, con agregados foliculares redondeados de manera concéntrica (en capa de cebolla) por linfocitos de pequeño tamaño, observándose a nivel central la presencia de pequeños vasos sanguíneos de paredes hialinizadas de manera prominente (fig. 2B). La conclusión fue EC tipo hialinovascular.
Se mantiene seguimiento de la paciente y 3 años después de su intervención se encuentra libre de enfermedad.
Discusión
La EC es una entidad poco frecuente, también denominada hiperplasia angiofolicular linfoide, hamartoma de ganglios linfáticos, linfoma gigante benigno, hiperplasia linfoide hialinizante, linforreticuloma folicular y angiomatosis linfoidea, lo que nos indica el desconocimiento acerca de su origen6,8. Esta afección puede encontrarse en el cuello (42%), mediastino (31%) y abdomen (23%), según Wang et al8, aunque hay otros autores que sitúan al mediastino como localización principal (70%)6.
Se desconoce su patogenia; sin embargo, se cree que hay una asociación con el virus de inmunodeficiencia humana y el virus herpes humano 8, debido a una anormal producción de interleucinas 6, que conlleva una linfoproliferación y la diferenciación de células plasmáticas6,8,9.
La EC de células plasmáticas afecta típicamente a más de un órgano, cursa habitualmente con síntomas generales (astenia, fiebre, mal estado general, hepatoesplenomegalia, etc.), hipergammaglobulinemia y aumento de la velocidad de sedimentación globular. Frecuentemente, se asocia a inmunodeficiencias, infecciones, sarcoma de Kaposi (13%), linfoma (18%), microangiopatías, carcinomas (colon, riñón y tiroides) y otras afecciones, como el síndrome POEMS (polineuropatía, organomegalia, endocrinopatía, proteinemia monoclonal y cambios en la piel)6,10. En el caso de nuestra primera paciente, se encontró aumento de la velocidad de sedimentación globular, hipergammaglobulinemia y asociación con el linfoma. Se presenta en cualquier grupo etario, con media en la quinta década de la vida, y al igual que el tipo hialinovascular, la mayoría de los autores informan que no hay un predominio de sexo; sin embargo, Wang et al indican un predominio en mujeres respecto a los varones de 2:18,10. En la TC se pueden encontrar adenopatías difusas y homogéneas en diferentes compartimientos mediastínicos, asociadas a esplenomegalia o hepatomegalia, como pudimos observar en el primer caso10. Desde un punto de vista histológico, en la variante plasnocelular, se observan extensas áreas de células plasmáticas maduras entre las estructuras foliculares transformadas, junto a la presencia de numerosos vasos sanguíneos, sin evidenciar vasos hialinizantes en los centros foliculares4,11.
Por otro lado, la EC hialinovascular, en contraste con el tipo plasmo celular, es una enfermedad asintomática, que se encuentra como hallazgo casual en las radiografías de tórax y, en caso de presentar síntomas, es por el efecto compresivo de la masa6,10. Se observa a cualquier edad, con predominio en la cuarta década de la vida. Aunque hay autores que afirman que afecta por igual a ambos sexos, otros informan de una prevalencia mayor en mujeres (4:1)10. En las pruebas de imagen, como la TC, se manifiesta como una masa homogénea o heterogénea que presenta calcificaciones entre el 5 y el 10% de los casos, y con la colocación de contraste se observa hipercaptación; aunque estos hallazgos no son diagnósticos. La RM muestra típicamente una imagen heterogénea hipointensa en T1 e hiperintensa en T210. Se caracteriza histológicamente por folículos con zonas muy hiperplásicas, formada por anillos celulares concéntricos de linfocitos pequeños (en capa de cebolla), dispuesto en torno a un centro germinal, con presencia de vasos hialinizados y células dendríticas prominentes; además, es diagnóstica la presencia de múltiples centros germinativos con estas características6,8.
Su diagnóstico preoperatorio es difícil, debido a que clínica y radiológicamente tiene características muy inespecíficas. La punción aspirativa con aguja fina usualmente no da el diagnóstico8.
En cuanto al tratamiento, en la forma multicéntrica, se han propuesto diferentes terapias, como combinación de radioterapia, quimioterapia y pautas de esteroides, además de la resección quirúrgica6,12. En la EC localizada, la exéresis completa quirúrgica es curativa en todos los casos. Algunos grupos emplean sistemáticamente embolización prequirúrgica, debido a la alta vascularización de este tumor, para evitar el riesgo de hemorragias6,8.
Nosotros hemos presentado dos casos de EC. El primer caso es el de una mujer joven, con clínica general y afectación de varios órganos, en el cual fue difícil llegar al diagnóstico, y en el que se plantearon como diagnóstico diferencial numerosas enfermedades hematológicas, como mieloma y linfoma. El segundo caso es el de una paciente con síntomas compresivos por la gran masa tumoral, la cual no presentó ningún signo de malignidad, excepto por su tamaño. Ambos casos se diferencian tanto en la presentación clínica como en su histología, pero en ambos el diagnóstico fue difícil, y fue necesario, tanto para la variante multicéntrica como para la localizada, la resección completa de la pieza para el diagnóstico definitivo.
Aunque el pronóstico en la EC diseminada es más incierto que en la variante localizada, en nuestros casos, la remisión de la enfermedad se ha conseguido por 8 y 3 años, respectivamente, con buena calidad de vida de ambas pacientes y sin recidiva de la enfermedad.
En conclusión, la EC es una rara enfermedad linfoproliferativa, de difícil diagnóstico, que debe tenerse en cuenta ante la presencia de adenopatías mediastínicas y/o masas torácicas, cuyo tratamiento es la resección tumoral con buen pronóstico y escasa evidencia de recidiva.
Conflicto de intereses
Los autores declaran que no tienen ningún conflicto de intereses.
*Autor para correspondencia.
Correo electrónico:karol.deaguiar@gmail.com (K. de Aguiar Quevedo).
Recibido el 23 de junio de 2011;
aceptado el 11 de julio de 2011