


Los sarcomas sinoviales son neoplasias de tejidos blandos, afectan a adultos jóvenes y de mediana edad, no existen diferencias con respecto a la distribución por sexo. Se localizan en el 90% de los casos en las extremidades.
El sarcoma sinovial pleuropulmonar primario (SSPP) es muy poco frecuente, constituye el 0,1% de los tumores pulmonares. Frecuentemente se manifiesta con tos y/o hemoptisis, el neumotórax es una forma poco común de presentación.
Son tumores con mal pronóstico y presentan alto riesgo de recidivas locales. Las mejores supervivencias están relacionadas con el tratamiento quirúrgico, siendo la resección completa tumoral el principal factor pronóstico. El tratamiento de elección es quirúrgico, seguido de quimioterapia, radioterapia o ambas.
Presentamos el caso clínico de un sarcoma pleuropulmonar primario que clínicamente se manifestó como neumotórax.
Synovial sarcomas are a soft-tissue neoplasms which affects young and middle-aged people, with no difference in distribution between sexes. They are located in the extremities in the 90% of the cases.
Pleuropulmonary primary synovial sarcoma SSPP is very unusual, constitutes 0.1% of pulmonary tumors. In most cases the SSPP present themselves with cough and / or hemoptysis whereas pneumothorax is an uncommon form of presentation.
Prognosis is usually poor, they present high risk of local recurrence. The survival rate depends on the surgical treatment, being the complete tumor resection the main predicting factor. Surgery is the treatment of choice followed by chemotherapy, radiotherapy or both.
We present a case report of a pleuropulmonary synovial sarcoma which presented with pneumothorax.
Introducción
El sarcoma sinovial pleuropulmonar (SSPP) es una entidad descrita en la literatura desde hace 15 años. Pertenece al grupo de los sarcomas sinoviales, tumores malignos de origen mesenquimal de células fusiformes caracterizado por una variable diferenciación epitelial, y citogenéticamente por una traslocación cromosomal específica t(X;18) (p11.2;q11.2)1. Debido a la similitud histológica es necesario la exclusión del sarcoma sinovial primario extratorácico para confirmar el diagnóstico de SSPP.
Son tumores agresivos, de evolución prolongada, con tendencia a la recidiva local. El tratamiento de elección es quirúrgico, el papel de la quimioterapia (QT) y la radioterapia (RT) aún no está bien definido.
Caso clínico
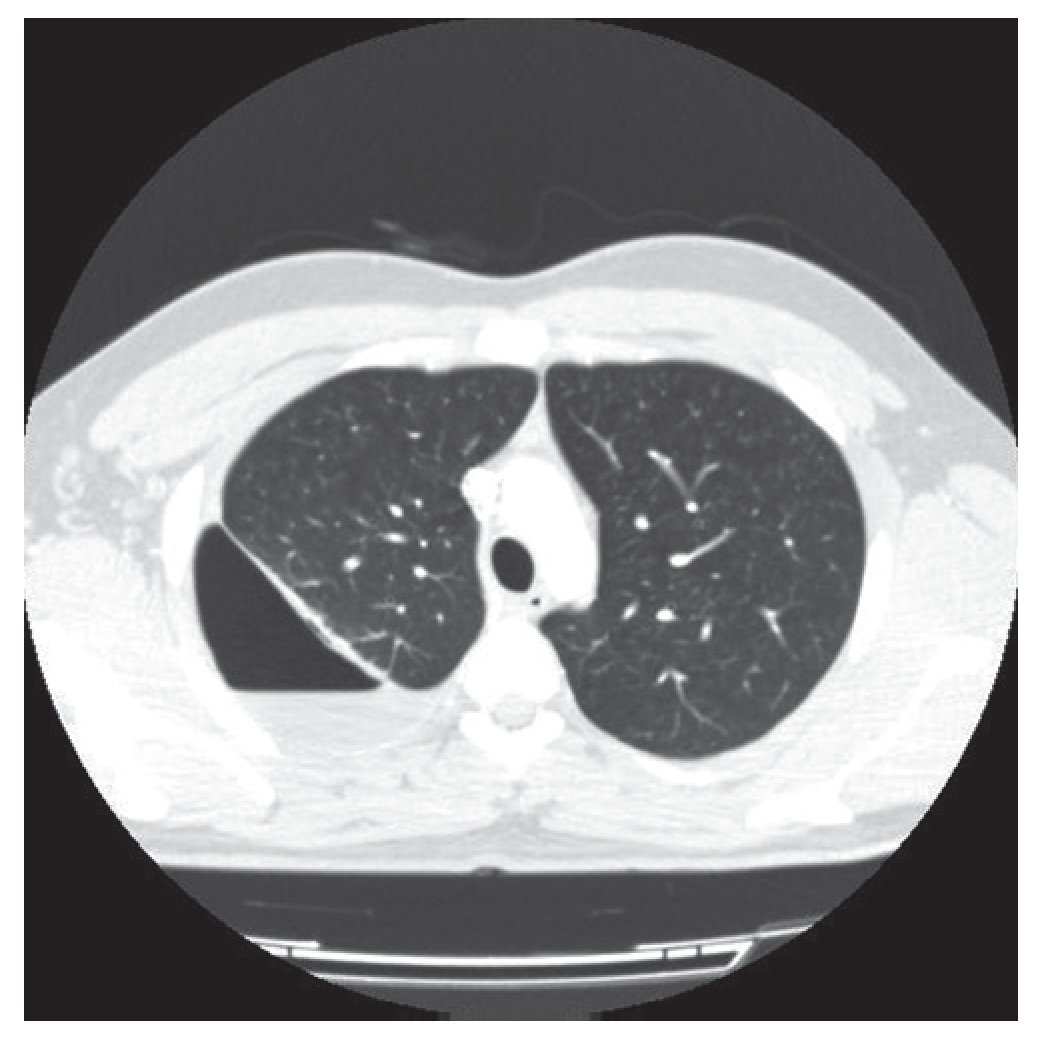
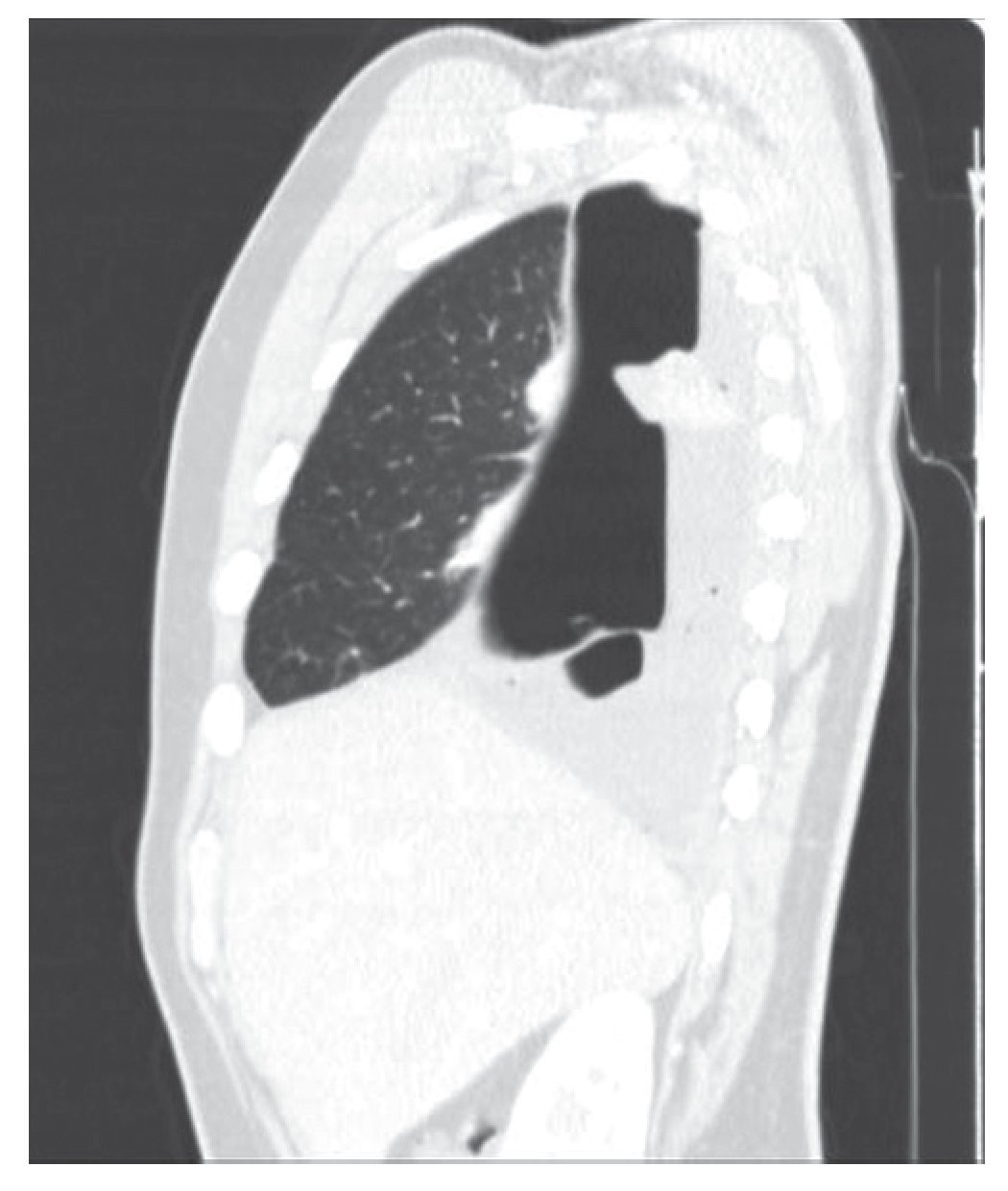
Varón de 21 años sin antecedentes médicos que acudió a Urgencias con un cuadro clínico y radiológico compatible con neumotórax derecho. El paciente refería en los meses previos, ocasionalmente y tras esfuerzos, disnea. Se decidió la colocación de un drenaje endotorácico y fue ingresado. Tras la obtención de la reexpansión pulmonar se procedió al alta hospitalaria con un seguimiento ambulatorio. Diez días después presentó de nuevo un cuadro clínico y radiológico compatible con hidroneumotórax que fue drenado de nuevo. Realizamos una tomografía computarizada (TC) torácica que reveló un hidroneumotórax derecho con una imagen densa pleuropulmonar (24 × 21 mm) captante de contraste, que retraía la cisura mayor hacia atrás (figs.1 y 2). No había signos sugestivos de lesión endobronquial. El pulmón izquierdo no presentaba alteraciones.
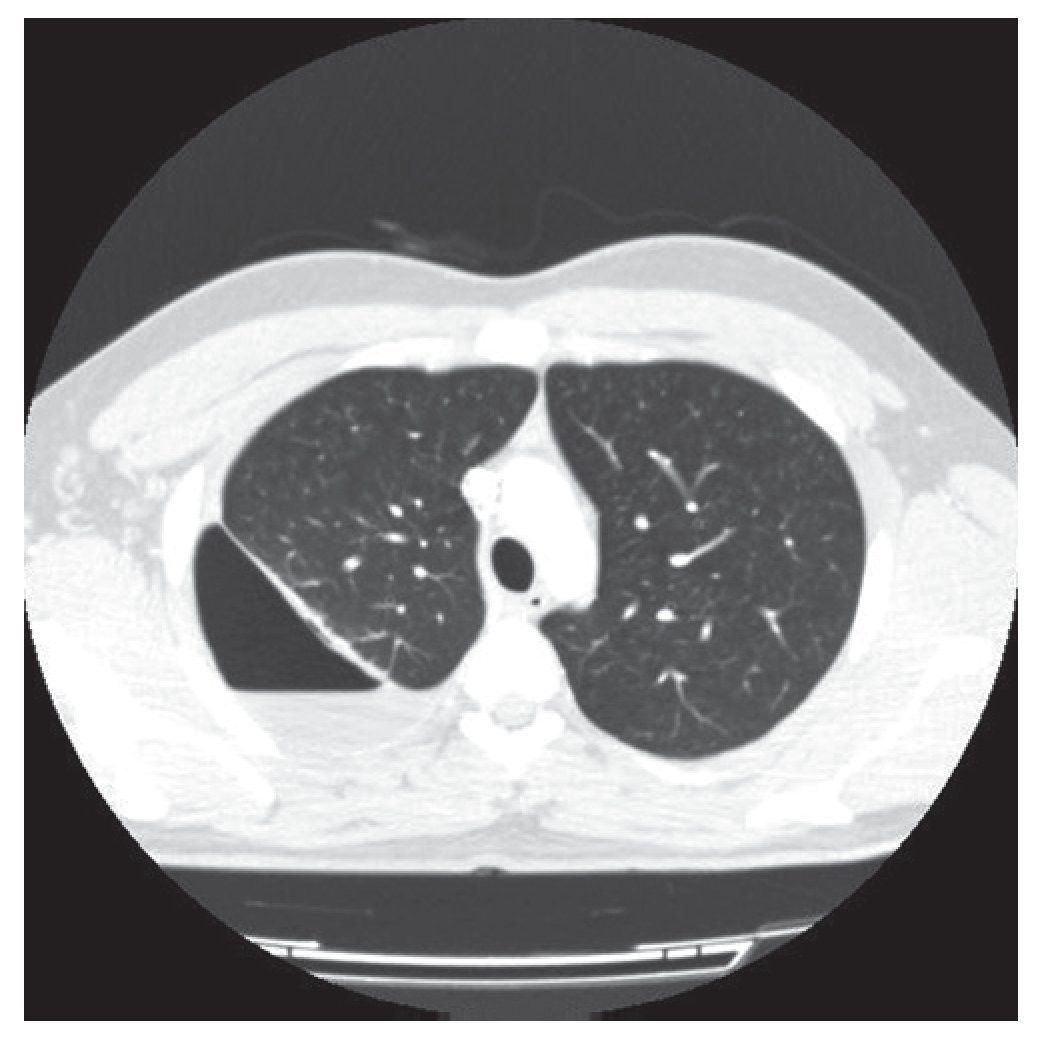
Figura 1 Hidroneumotórax derecho con una imagen densa pleuropulmonar (24 × 21 mm).
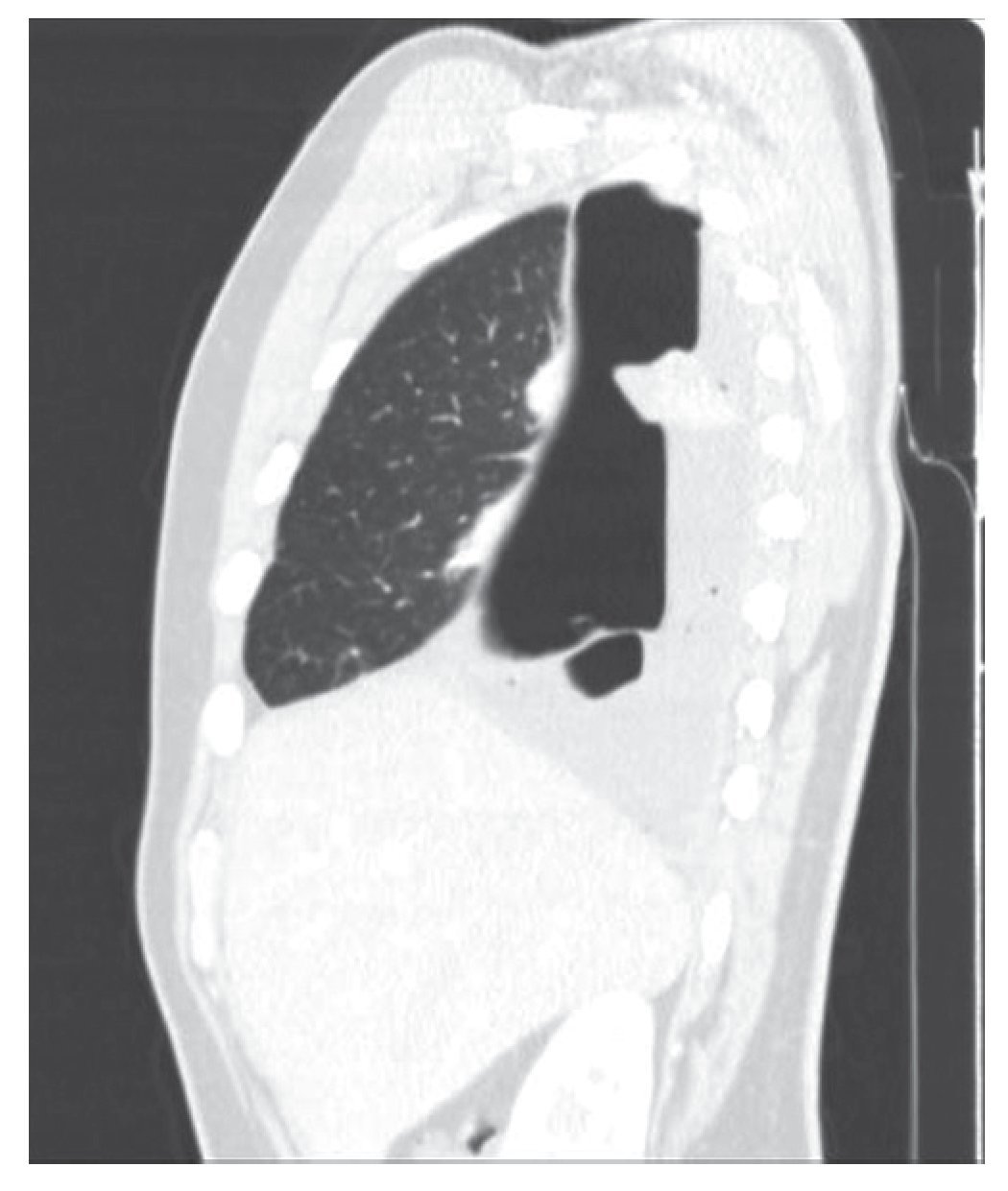
Figura 2 Imagen que retraía la cisura mayor hacia atrás.
Ante estos hallazgos se decidió la realización de cirugía videotoracoscopia VATS y biopsia pleuropulmonar. Visualizamos un pulmón adherido por múltiples adherencias laxas, engrosamineto pleuroparietal y nódulo en el lóbulo superior derecho (LSD) adherido a pleura parietal. El resultado del estudio anatomopatológico fue de sarcoma sinovial primario monofásico. Dado el diagnóstico se realizó una nueva anamnesis, no refería dolor articular, y ampliamos el estudio con una tomografía de emisión de positrones (PET) que mostró hipercaptación tan sólo a nivel del nódulo pulmonar y pleura parietal donde estaba adherido. De forma programa realizamos una lobectomía superior derecha y pleurectomía parietal de la zona apicoposterior.
En el estudio anatomopatológico de la pieza con técnicas inmunohistoquímicas se observó positividad intensa y difusa con vimentina, bcl-2 y CK policlonal. Positividad focal con CK-7, EMA y calretinina. Tinción negativa con S-100, Actina, TTF-1, CEA policlonal, desmina, CD-34 y CK-20. Los resultados inmunohistoquímicos fueron compatibles con el diagnóstico de sarcoma sinovial monofásico. Se realizó la determinación de la traslocación (X;18) que confirmó el diagnóstico.
Tras la cirugía, el paciente recibió QT, regímenes de isofosfamida y doxorubicina, y RT adyuvantes.
Dos años después, el paciente permanece libre de enfermedad.
Discusión
Los sarcomas sinoviales representan entre el 3-10% de todos los sarcomas2. Es una neoplasia de tejidos blandos originada no de tejido sinovial, sino de células mesenquimales pluripotenciales3. En el 90% de los casos se sitúa a nivel para-articular en las extremidades, con frecuencia en las inferiores.
La exclusión de sarcoma sinovial primario extratorácico es esencial para confirmar el diagnóstico de SSPP.
El SSPP es poco frecuente, constituye el 0,1% de todos los tumores de pulmón. Afecta a varones jóvenes y de mediana edad y no existe diferencia de distribución con respecto al sexo4.
Histológicamente existen tres variantes, la monofásica propiamente dicha (células fusiformes y raramente epiteliales), una variante epitelial monofásica y la bifásica (células fusiformes y epiteliales). El diagnóstico diferencial se plantea, entre otros, con carcinoma sarcomatoide, blastoma pulmonar, mesotelioma, tumor fibroso solitario, fibrosarcoma, leiomiosarcoma y metástasis, por lo que para su diferenciación se hace necesario el empleo de técnicas inmunohistoquímicas y ultraestructurales. Las células fusiformes y en menor medida las epiteliales se marcan con vimentina, que es un marcador mesenquimal. Algunos sarcomas sinoviales expresan proteína S-100, en nuestro caso esta fue negativa. Es necesario para su diagnóstico que un marcador epitelal sea positivo.
Genéticamente se ha demostrado que en el 95% de los casos de sarcoma sinovial existe una traslocación recíproca específica en el cromosoma (X y 18) (p11.2, q11.2), esta es el resultado de la fusión del gen SYT del cromosoma 18 con el gen SSX1 o SSX2 del cromosoma X. El 92% de los SSPP son positivos para t(X; 18)5.
Suelen ser lesiones únicas, de crecimiento lento, sin preferencia por ningún lóbulo pulmonar. La localización endobronquial es infrecuente.
La clínica más frecuente es la tos y la hemoptisis, esta última debida a la presencia de formaciones quísticas y lesiones necrohemorrágicas del tumor. El 25% de los casos son asintomáticos. En nuestro caso el tumor se manifestó con clínica de neumotórax, forma poco común de presentación.
Radiológicamente, mediante la realización de una TC torácica podemos observar masas pulmonares de implantación pleuropulmonar con áreas de necrosis y componentes de tejidos blandos. Es frecuente la presencia de derrame pleural ipsilateral y rara la afectación de adenopatías mediastínicas.
El tratamiento es quirúrgico, tanto de la lesión primaria como de las metástasis. Se deben realizar resecciones económicas debido al alto riesgo de recidiva local, hasta un 75% según algunas series6.
La QT y la RT adyuvante pueden reducir y retrasar la aparición de metástasis a distancia. Actualmente se está tratando a los pacientes mediante cirugía seguida de QT y RT.
En pacientes irresecables o avanzados la QT y RT podrían ser de utilidad. La respuesta a la QT es moderada, entorno al 50% de los casos, con regímenes de isofosfamida y doxorubicina7.
El pronóstico es pobre, los sarcomas tienen una evolución prolongada, algunos pacientes sobreviven hasta 20 años; sin embargo la media de supervivencia es de 5-6 años. Las mejores supervivencias están relacionadas con el tratamiento quirúrgico. El SSPP es más agresivo que el localizado en partes blandas, con una media de supervivencia de 50 meses y del 30 al 76% a los 5 años según las series8.
El tamaño tumoral (> 9 cm), el sexo masculino, la edad (> 20), la necrosis extensa del tumor, la alta actividad miótica (> 9-10 mitosis por campo), la invasión neurovascular y la variante SYT-SSX1 son los factores que indican peor pronóstico9.
El principal factor pronóstico es la obtención o no obtención completa de la resección tumoral.
Conflicto de intereses
Los autores declaran que no tienen ningún conflicto de intereses.
* Autor para correspondencia.
Correo electrónico: mirissima@hotmail.com
Recibido el 13 de septiembre de 2011;
aceptado el 16 de mayo de 2012