







Revisar o atendimento integral atual de craniossinostose não sindrômica e deformidade craniana não sinostótica e oferecer uma visão global dessas condições craniofaciais.
Fontes de dadosA revisão foi feita nas bases de dados PubMed, SciELO e Lilacs e sem restrições de tempo ou idioma. Artigos relevantes foram selecionados para a revisão.
Síntese dos dadosForam incluídas a anatomia e a fisiologia do desenvolvimento normal do crânio em crianças, discutidas nuances relacionadas à nomenclatura, epidemiologia, etiologia e ao tratamento das formas mais comuns de craniossinostose não sindrômica. Também foram discutidos os critérios clínicos para o diagnóstico diferencial entre deformidades posicionais e craniossinostose não sindrômica. Deram‐se aos pediatras subsídios para um diagnóstico clínico rápido e seguro. Se deformidades posicionais forem diagnosticadas com precisão, elas podem ser tratadas com sucesso por meio da modificação do comportamento. Dúvidas de diagnóstico e pacientes portadores de craniossinostose devem ser encaminhados imediatamente a um centro multidisciplinar craniofacial.
ConclusõesOs pediatras estão na vanguarda do diagnóstico de pacientes com deformidades cranianas. Assim, é de suma importância que reconheçam deformidades cranianas sutis, pois elas podem estar relacionadas à fusão prematura das suturas cranianas.
To review the current comprehensive care for nonsyndromic craniosynostosis and nonsynostotic cranial deformity and to offer an overall view of these craniofacial conditions.
Data sourceThe review was conducted in the PubMed, SciELO, and LILACS databases without time or language restrictions. Relevant articles were selected for the review.
Data synthesisWe included the anatomy and physiology of normal skull development of children, discussing nuances related to nomenclature, epidemiology, etiology, and treatment of the most common forms of nonsyndromic craniosynostosis. The clinical criteria for the differential diagnosis between positional deformities and nonsyndromic craniosynostosis were also discussed, giving to the pediatrician subsidies for a quick and safe clinical diagnosis. If positional deformity is accurately diagnosed, it can be treated successfully with behavior modification. Diagnostic doubts and craniosynostosis patients should be referred straightaway to a multidisciplinary craniofacial center.
ConclusionsPediatricians are in the forefront of the diagnosis of patients with cranial deformities. Thus, it is of paramount importance that they recognize subtle cranial deformities as it may be related to premature fusion of cranial sutures.
Deformidades cranianas são queixas comuns em unidades pediátricas, já que 25% das crianças de gestações únicas e 50% das de gravidezes múltiplas têm algum grau de deformidade no crânio ao nascimento. Em geral, os pais geralmente reconhecem essas alterações nas primeiras semanas ou nos primeiros meses de vida.1 No entanto, em alguns cenários, o diagnóstico pode ser negligenciado pela família, que tende a negar o problema. Nesses casos, os pediatras devem estar cientes dessas questões e aconselhar a família sobre a importância de procurar uma equipe craniofacial. Além disso, é de fundamental importância que o pediatra esteja preparado na primeira consulta para fazer o diagnóstico diferencial entre uma deformidade posicional e craniossinostose, considerando‐se que as crianças que nascem com uma deformidade posicional não precisam ser expostas à radiação ionizante de uma tomografia computadorizada (CT), além de considerarmos os custos do procedimento e os riscos da sedação para obtê‐lo.2
Neste relato, revisamos a anatomia e a fisiologia do desenvolvimento craniano normal de crianças, discutimos as nuances relacionadas à nomenclatura, epidemiologia, etiologia e ao tratamento das formas mais comuns de craniossinostose. Os critérios clínicos para o diagnóstico diferencial entre deformidades posicionais e craniossinostose também são apresentados, o que possibilita subsídios ao pediatra para um diagnóstico clínico rápido e seguro.
MétodoO presente estudo é uma revisão da literatura com abordagem descritiva. A revisão da literatura foi feita por meio de busca nas bases de dados Medline (PubMed), SciELO e Lilacs, sem restrições de tempo ou de idioma. A revisão final da literatura foi feita em julho de 2015. Para identificar todos os artigos relevantes (artigos de revisão, ensaios clínicos e estudos de coorte) sobre os cuidados abrangentes atuais para craniossinostoses não sindrômicas e deformidades cranianas não sinostóticas, os seguintes termos de pesquisa foram usados: “craniossinostose não sindrômica”, “deformidade craniana não sinostótica”, “deformidade posicional”, “plagiocefalia posterior não sinostótica” e “plagiocefalia posicional”. Cada estudo relevante foi revisado individualmente para identificar informações sobre o desenvolvimento normal do crânio em crianças, a nomenclatura, epidemiologia, etiologia, o diagnóstico e tratamento das formas mais comuns de craniossinostose não sindrômica e deformidade craniana não sinostótica.
Anatomia cranianaO crânio de um recém‐nascido é composto de múltiplos ossos e suturas que, para permitir sua passagem através do canal de parto e acomodar o encéfalo, o tornam maleável e sujeito a forças externas que o deformam, visto que o volume do cérebro é quadruplicado nos dois primeiros anos de vida.3
O crânio é composto de quatro suturas primárias (metópica, sagital, coronal e lambdoide), três suturas secundárias (frontonasal, escamosa temporal e frontoesfenoidal) e quatro ossos principais (temporal, frontal, parietal e occipital). A sutura metópica separa os ossos frontais uns dos outros, a sutura sagital separa os ossos parietais, a sutura coronal separa os ossos parietal e frontal e a sutura lambdoide separa os ossos parietal e occipital.3 Além dos ossos e das suturas, um espaço mole e membranoso, denominado fontanela, que separa os ossos do crânio, é de grande importância e tem a fontanela anterior ou bregmática (delimitada pelos ossos frontal e parietal) e a fontanela posterior ou lambdoide (delimitada pelos ossos occipital e parietal).
Em condições fisiológicas, as suturas cranianas progridem para a fusão com diferentes períodos iniciais de fusão, de acordo com cada uma das suturas primárias (tabela 1). O mesmo ocorre com as fontanelas, que geralmente se fecham até o segundo ano (tabela 2).
Suturas cranianas primárias e secundárias e idade no início da fusão3
| Suturas | Início da fusão |
|---|---|
| Metópica | 2 meses |
| Sagital | 22 meses |
| Coronal | 24 meses |
| Lambdoide | 26 meses |
| Frontonasal | 68 meses |
| Frontoesfenoidal | 22 meses |
| Escamosa temporal | 35‐39 meses |
Idade de fechamento das fontanelas cranianas3
| Fontanelas | Idade de fechamento |
|---|---|
| Anterior ou bregmática | 24 meses |
| Posterior ou lambdoide | 3 meses |
| Anterolateral (Esfenoide) | 6‐24 meses |
| Posterolateral (Mastoide) | 6‐24 meses |
Um detalhe importante é que os ossos do crânio são membranosos (sem uma fase cartilaginosa anterior), o que resulta em crescimento por meio de deposição óssea na região da sutura; esse crescimento ocorre perpendicular à sutura. Por exemplo, as suturas coronais permitem o crescimento anteroposterior do crânio, enquanto a sutura sagital permite o crescimento do crânio biparietal.3
CraniossinostoseA definição de craniossinostose é a fusão prematura de uma ou mais suturas cranianas. A ocorrência é de aproximadamente uma em 2.000‐2.500 nascidos vivos.4 A fusão prematura das suturas impede o crescimento perpendicular do crânio e um aumento no volume do cérebro leva a um crescimento compensatório do crânio paralelo a esse aumento.
As craniossinostoses são classificadas como primária ou secundária. A craniossinostose primária resulta de influências genéticas e ambientais, é classificada como simples e complexa. A craniossinostose complexa é adicionalmente dividida em não sindrômica e sindrômica (tabela 3).5–8 Devido à maior prevalência, discutiremos o diagnóstico de craniossinostose simples primária; isto é, as sinostoses sagital, coronal, metópica e lambdoide (fig. 1). Subsequentemente, discutiremos a plagiocefalia posterior não sinostótica (plagiocefalia posicional) e enfatizaremos características importantes para o diagnóstico diferencial com sinostose lambdoide (plagiocefalia posterior).
Classificação de craniossinostoses
| 1. Primária |
| Simples (envolve uma única sutura): sagital, coronal, metópica e lambdoide |
| Complexa (fusão de duas ou mais suturas) |
| Não sindrômica: bicoronal |
| Sindrômica: Crouzon, Apert, Pfeiffer e Saethre‐Chotzen |
| 2. Secundária |
| Distúrbios metabólicos: hipertireoidismo, erros inatos do metabolismo |
| Várias malformações: microcefalia, encefalocele |
| Após derivação ventricular com drenagem excessiva de líquido cefalorraquidiano |
| Exposição fetal a determinadas substâncias: ácido valproico, fenitoína |
| Mucopolissacaridose |
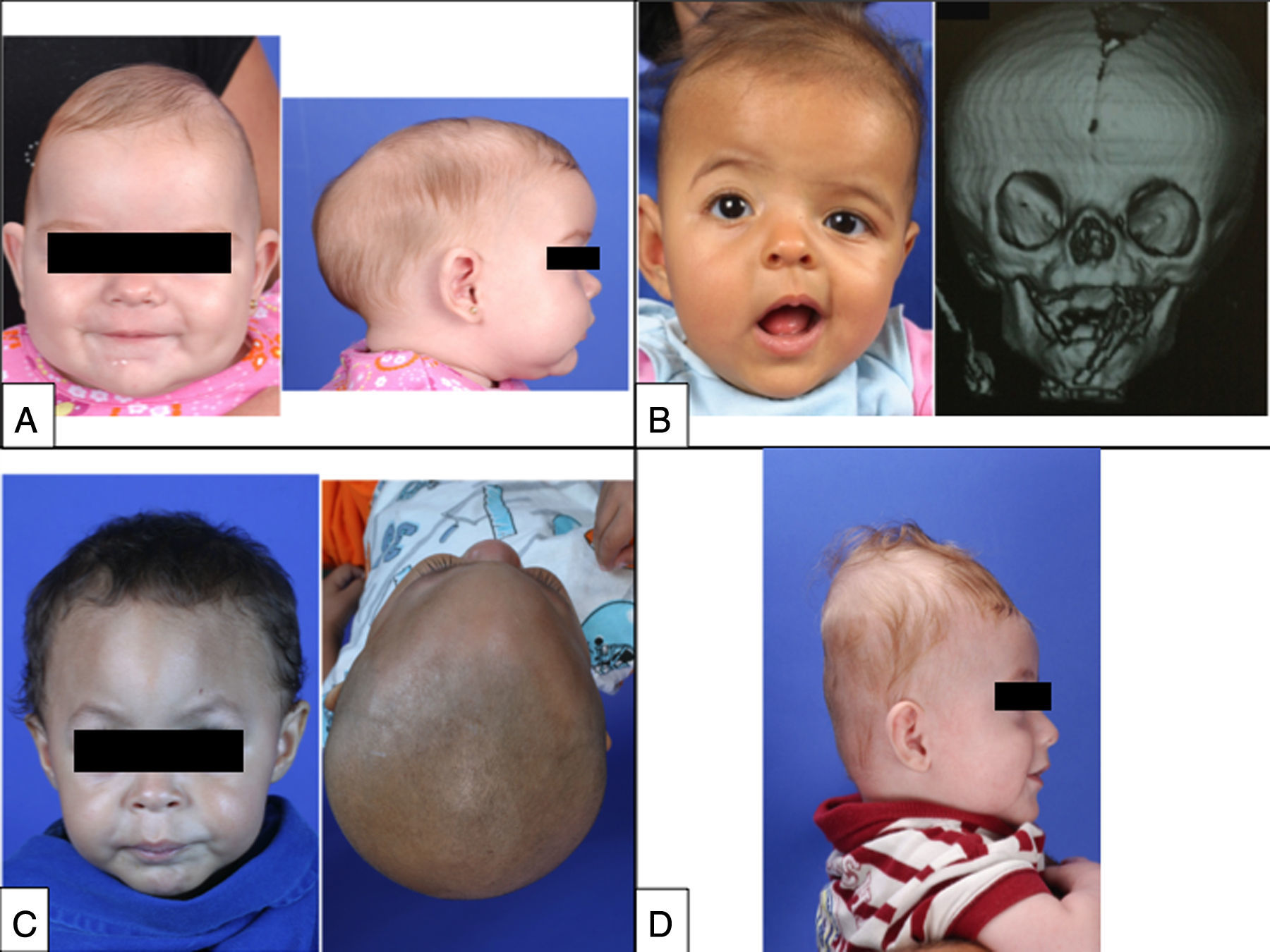
 Fotografia frontal de paciente com fusão prematura da sutura sagital que mostra a constrição temporal característica. (Direita) Fotografia lateral que mostra aumento no diâmetro anteroposterior do crânio (crânio longo e estreito), bossa frontal e protuberância occipital, que são as principais características clínicas de craniossinostose sagital. (B, Esquerda) Fotografia frontal de paciente com fusão prematura da sutura coronal direita que mostra a retrusão da fusão óssea frontal ipsilateral e o abaulamento contralateral compensatório, a assimetria das sobrancelhas, órbitas, orelhas, do nariz e da mandíbula, bem como estrabismo convergente do olho esquerdo. (Direita) Reconstrução de TC em 3D que mostra a fusão prematura da sutura coronal direita e a elevação da asa do esfenoide ipsilateral que levam a uma órbita alongada, reconhecida como “olhar de arlequim”. (C, Esquerda) Fotografia frontal de paciente com fusão prematura da sutura metópica que mostra o aspecto triangular da testa com retrusão das cristas orbitais bilateralmente e hipoteleorbitismo (aproximação das órbitas). (Direita) Vista basal que revela a aparência triangular do crânio. (D) Fotografia lateral de paciente com fusão prematura da sutura lambdoide que mostra o aspecto turricefálico do crânio. As fotografias bidimensionais e as documentações radiológicas pertencem aos arquivos do Hospital Sobrapar. Os termos de consentimento informado foram assinados pelos pais dos pacientes.")
(A, Esquerda) Fotografia frontal de paciente com fusão prematura da sutura sagital que mostra a constrição temporal característica. (Direita) Fotografia lateral que mostra aumento no diâmetro anteroposterior do crânio (crânio longo e estreito), bossa frontal e protuberância occipital, que são as principais características clínicas de craniossinostose sagital. (B, Esquerda) Fotografia frontal de paciente com fusão prematura da sutura coronal direita que mostra a retrusão da fusão óssea frontal ipsilateral e o abaulamento contralateral compensatório, a assimetria das sobrancelhas, órbitas, orelhas, do nariz e da mandíbula, bem como estrabismo convergente do olho esquerdo. (Direita) Reconstrução de TC em 3D que mostra a fusão prematura da sutura coronal direita e a elevação da asa do esfenoide ipsilateral que levam a uma órbita alongada, reconhecida como “olhar de arlequim”. (C, Esquerda) Fotografia frontal de paciente com fusão prematura da sutura metópica que mostra o aspecto triangular da testa com retrusão das cristas orbitais bilateralmente e hipoteleorbitismo (aproximação das órbitas). (Direita) Vista basal que revela a aparência triangular do crânio. (D) Fotografia lateral de paciente com fusão prematura da sutura lambdoide que mostra o aspecto turricefálico do crânio. As fotografias bidimensionais e as documentações radiológicas pertencem aos arquivos do Hospital Sobrapar. Os termos de consentimento informado foram assinados pelos pais dos pacientes.
Várias teorias foram propostas para explicar a patogênese de uma fusão anormal de sutura craniana. Fatores tanto ambientais (especialmente constrição intrauterina da cabeça fetal) quanto genéticos (mutações de um único gene, anormalidades cromossômicas e componente poligênico) predispõem à craniossinostose.9–11 Mutações em sete genes (FGFR1, FGFR2, FGFR3, TWIST1, EFNB1, MSX2 e RAB23) são inequivocamente associadas a formas mendelianas de craniossinostose.9–11 Em contraste, a etiologia genética da craniossinostose não sindrômica permaneceu mal compreendida até muito recentemente.9–11 Nos últimos anos, estudos epidemiológicos e fenotípicos demonstram claramente que a craniossinostose não sindrômica é uma condição complexa e heterogênea sustentada por um forte componente genético acompanhado por fatores ambientais que contribuem para a rede da patogenia desse defeito congênito.9,10 De fato, mutações raras em FGFRs, TWIST1, LRIT3, ALX4, IGFR1, EFNA4, RUNx2 e FREM1 foram relatadas em uma pequena fração de pacientes com craniossinostose não sindrômica.9,10 Os testes genéticos moleculares mínimos recomendados para cada apresentação clínica (craniossinostose sindrômica e não sindrômica) foram previamente revistos em uma publicação10 e estão fora do escopo deste relato. Pesquisas adicionais em grandes populações com subconjuntos de pacientes fenotipicamente homogêneos são necessárias para compreender os fatores genéticos, maternos, ambientais e estocásticos complexos que contribuem para a craniossinostose não sindrômica.9–11
Abordagem geral para o diagnósticoEspera‐se que os pediatras consigam reconhecer as deformidades cranianas e diagnosticá‐las como craniossinostoses ou deformidades cranianas posicionais. Se uma deformidade craniana estiver presente, o exame físico e a anamnese (principais características descritas nos próximos subtítulos) são as peças mais úteis e reveladoras de informação na avaliação da criança. Um fluxograma anamnésico publicado anteriormente12 serve como um guia para distinguir a craniossinostose de deformidades cranianas posicionais. As perguntas mais importantes para diferenciar as craniossinostoses das deformidades não sinostóticas são: 1) “A deformidade estava presente no nascimento?” A craniossinostose está presente no nascimento, enquanto as deformidades não sinostóticas se desenvolvem no período neonatal; 2) “Existe uma posição preferida para dormir?”; 3) “Houve melhoria da deformidade?” A craniossinostose piora com o tempo, enquanto as deformidades não sinostóticas melhoram à medida que a criança desenvolve o controle da cabeça e o crânio não está mais sob pressão localizada por longos períodos.12
Em casos raros e difíceis, quando o exame e a anamnese não são diagnósticos, uma série radiográfica de boa qualidade de quatro incidências (anteroposterior, Towne e duas projeções laterais) pode ser suficiente para excluir craniossinostose e evitar uma exposição maior à radiação.13 Caso esteja obscura devido à pouca idade do paciente, recomenda‐se repetir a radiografia craniana após um ou dois meses.14 TC não é a modalidade recomendada para o rastreamento em função da exposição à radiação e do alto custo associados e o diagnóstico com TC feito por pediatras está associado à demora adicional no encaminhamento.13,14
Após a suspeita clínica (ou confirmação) de craniossinostose, a criança deve ser encaminhada a uma equipe multidisciplinar especializada em anomalias craniofaciais (anestesista, cirurgião plástico, fonoaudiólogo, neurocirurgião, ortodontista, otorrinolaringologista e psicólogo).15 Nesses centros, o exame radiológico de escolha é a TC tridimensional que contribui para elucidar a extensão da fusão sutural e a consequente deformidade craniofacial e o planejamento cirúrgico subsequente. Vale ressaltar que o perímetro cefálico geralmente não muda devido ao crescimento compensatório de outros ossos na maioria dos casos com craniossinostose simples.
Nesse contexto, os pacientes com craniossinostose não tratados cirurgicamente podem desenvolver várias complicações,16–18 como hipertensão intracraniana (HIC), que ocorre em até 60% das crianças com craniossinostose complexa e 20% dos portadores de craniossinostose simples; distúrbios cognitivos e de desenvolvimento como ganho insuficiente de peso; distúrbios visuais, de audição e de linguagem e problemas psicológicos, como autoestima baixa e isolamento social. Portanto, o objetivo do tratamento cirúrgico é evitar a HIC e corrigir as anomalias craniofaciais. Em geral, o momento ideal para a correção cirúrgica é entre 6‐9 meses na maioria dos casos. As motivações para a cirurgia antes de um ano de idade incluem a capacidade de reossificação completa da criança com menos de um ano, o caráter maleável da calota craniana nessa idade e o enorme crescimento cerebral que ocorre durante o primeiro ano de vida, o que permite a boa remodelação do crânio.19 O formato craniofacial satisfatório e resultados estéticos agradáveis também foram associados a intervenções cirúrgicas craniofaciais feitas antes de um ano de idade.18,19 Vale ressaltar que a presença de sinais de HIC (irritabilidade, edema papilar, abaulamento da fontanela e achados radiológicos) pode resultar na necessidade de intervenção cirúrgica precoce para procedimentos de descompressão ou cirurgia de derivação ventricular, se associados à hidrocefalia.
Sinostose sagitalA sinostose sagital é a forma mais comum de craniossinostose simples, responsável por 40‐60% dos casos, é mais prevalente no sexo masculino (75‐85%). O formato do crânio é alongado e estreito, semelhante a um barco, motivo pelo qual é denominado escafocefalia (fig. 1A).20 Ao exame físico, um cume pode ser palpado na sutura sagital. Deve‐se notar que a fontanela anterior pode não estar fechada. Uma bossa frontal compensatória e protrusão occipital podem ocorrer em graus variados.
O tratamento cirúrgico é indicado entre 3‐12 meses de idade e os procedimentos podem variar de uma simples ressecção endoscópica da sutura sagital a uma reconstrução total do crânio, a depender da gravidade do estado clínico. Em nosso serviço, recomendamos a cirurgia entre 6‐9 meses e o uso de craniectomia em formato da letra grega “π” (denominado procedimento Hung Span),21 associado a várias osteotomias (cortes ósseos), retângulos paralelos de cerca de dois centímetros de comprimento no osso parietal, entre as suturas coronal e lambdoide, o que permite maior espaço lateral para acomodação adicional do cérebro. Osteotomias em formato de aduelas de barril (cortes ósseos laterais) ainda permitem a redução do diâmetro anteroposterior e melhor remodelação do crânio, com resultados estéticos excelentes.22
Sinostose coronalA sinostose coronal é a segunda forma mais comum, responsável por até 25% dos casos de craniossinostose. O fechamento de uma sutura coronal é denominado plagiocefalia anterior,23,24 enquanto o encerramento de duas suturas é denominado braquicefalia (comumente encontrada em craniossinostose sindrômica).6–8 Afeta predominantemente o sexo feminino (60%), com incidência semelhante em ambos os lados.
A fusão precoce da sutura coronal leva a um achatamento do osso frontal e do aro orbital ipsilateral à fusão, com uma bossa frontal compensatória contralateral.23,24 O estrabismo é um achado comum (50‐60% dos casos) e resulta de alterações morfológicas no teto orbitário e na tróclea, altera a função do músculo oblíquo superior.25 A elevação da asa do esfenoide ipsilateral pode ser observada em radiografia simples do crânio e é reconhecida como “olho de arlequim” (fig. 1B).26 A fusão prematura da sutura coronal também provoca um desvio da base do crânio, altera a posição das órbitas e causa assimetria das sobrancelhas e da posição das orelhas, desvio da mandíbula e má oclusão, com um efeito estético importante.23,24
Portanto, o tratamento cirúrgico é indicado para correção da deformidade morfológica do crânio e as suas repercussões sobre a face e também por causa do estrabismo e dos riscos de desenvolver HIC. Recomendamos o procedimento entre 6‐9 meses, quando já existe maturidade óssea suficiente para a remodelação. Basicamente, um avanço fronto‐orbital associado ao remodelamento frontal é feito, libera ambas as suturas coronais.24 Procedimentos adicionais, como injeção de gordura no esqueleto craniofacial para diminuir assimetrias faciais e correção de ptose palpebral, podem ser necessários. No fim do crescimento facial, cirurgia ortognática e rinoplastia com septoplastia podem ser feitas por um cirurgião plástico com formação na área de cirurgia craniofacial.
Sinostose metópicaA sinostose metópica corresponde a 10% de todas as craniossinostoses e predomina no sexo masculino (75‐85% dos casos). A fusão prematura da sutura metópica restringe o crescimento transversal dos ossos frontais e, em casos mais graves, pode restringir a expansão da fossa anterior, o que leva ao hipoteleorbitismo e, consequentemente, à trigonocefalia (fig. 1C). A craniossinostose metópica é a única sinostose sutural comumente associada a distúrbios cognitivos, principalmente devido à restrição do crescimento dos lobos frontais.27
O aumento no volume da fossa anterior é o principal objetivo no tratamento de pacientes com trigonocefalia, bem como a remodelação frontal e o avanço fronto‐orbital. A melhor época para o tratamento é entre 6‐9 meses de idade.
Sinostose lambdoideA sinostose lambdoide é a forma mais rara de craniossinostose simples, com uma incidência de aproximadamente 3/10.000 nascidos vivos, corresponde a cerca de 1,0‐5,5% de todas as craniossinostoses. Quando avaliada em uma população de crianças com achatamento occipital (também denominado plagiocefalia posterior), é responsável por apenas 0,9‐4% dos casos.28
A fusão de uma sutura lambdoide leva a uma deformidade occipital (plagiocefalia posterior) com um formato trapezoidal, enquanto a fusão de ambas as suturas lambdoides leva à braquicefalia (fig. 1D). Além da deformidade posterior (achatamento, mau posicionamento das orelhas, bossa parietal compensatória) causada pela fusão prematura da sutura lambdoide, alterações morfológicas significativas podem ocorrer concomitantemente na fossa posterior. Essa craniossinostose está associada a tonsilas cerebelares herniadas (também conhecidas como malformação de Chiari do tipo I) e à fusão do forame jugular, resultam em um alto risco de hipertensão venosa. Logo, essa é a forma de craniossinostose simples com o maior risco de HIC.29
O tratamento cirúrgico tem como base a expansão do volume da porção posterior do crânio (região parietal e occipital) e a liberação das suturas lambdoides. Porém, essa região tem grande drenagem venosa, com numerosas veias do couro cabeludo que atravessam o osso em direção aos seios durais, aumentam imensamente o risco cirúrgico de uma craniotomia e remodelação óssea. Em nosso serviço, optamos pela técnica de distração posterior, na qual o volume craniano é gradualmente aumentado, o que reduz significativamente o risco de sangramento e a necessidade de transfusões sanguíneas.30
Plagiocefalia posicionalO termo plagiocefalia significa crânio oblíquo e corresponde a um achatamento occipital unilateral ou bilateral, que pode surgir devido à influência contínua de forças externas sobre o crânio imaturo (plagiocefalia sinostótica posterior) ou devido à fusão prematura de uma ou de ambas as suturas lambdoides (plagiocefalia sinostótica posterior). A plagiocefalia anterior pode ser usada para definir a deformação craniana caracterizada por fusão unilateral prematura da sutura coronal.
A plagiocefalia posicional ou deformacional é a causa mais comum de plagiocefalia (prevalência de 5‐48% em recém‐nascidos saudáveis)31versus uma incidência de 0,003% de plagiocefalia sinostótica (sinostose lambdoide).28
Com base na introdução da campanha de prevenção da síndrome da morte súbita do lactante (programa Back to Sleep) no início da década de 1990 – recomenda que os bebês permaneçam em posição supina –, um aumento significativo na incidência de crianças com plagiocefalia posicional foi observado (5‐48%).31 Essa deformidade resulta de uma ação permanente das forças gravitacionais na região occipital, causa uma região achatada no esqueleto craniofacial posterior. Se nenhuma intervenção for feita, a deformidade pode continuar e, em casos graves, evoluir com deformidades faciais. A plagiocefalia posicional ocorre com mais frequência do lado direito (70% dos casos) e afeta mais o sexo masculino. Os principais fatores de risco incluem torcicolo, prematuridade, multiparidade e uma posição de dormir fixa.
O diagnóstico é eminentemente clínico e a diferenciação com plagiocefalia sinostótica (fusão unilateral das suturas coronal ou lambdoide) é essencial.28 Anamnese e exame físico são suficientes para estabelecer o diagnóstico diferencial entre uma deformidade posicional e craniossinostose, na grande maioria dos casos (tabela 4). Classicamente, os pacientes com fusão prematura da sutura lambdoide já apresentam a deformidade ao nascer, enquanto aqueles com uma deformidade posicional apresentam um crânio normal ao nascimento e desenvolvem a deformidade nas semanas ou meses seguintes. Quando questionados, os pais podem mencionar que há uma posição preferida de posicionamento do bebê em pacientes com plagiocefalia posicional, enquanto em pacientes com plagiocefalia sinostótica não há posição preferida.
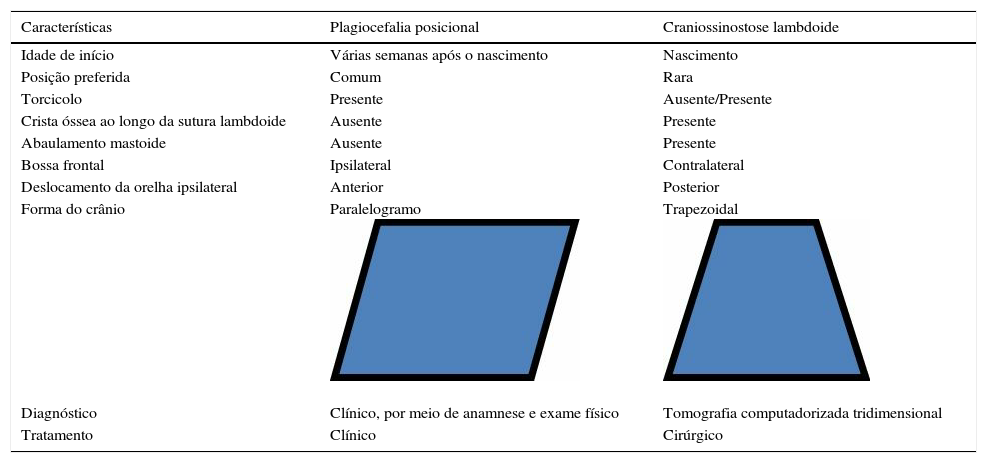
Características importantes para subsidiar o diagnóstico diferencial de plagiocefalia posicional versus sinostose lambdoide21,24,25
| Características | Plagiocefalia posicional | Craniossinostose lambdoide |
|---|---|---|
| Idade de início | Várias semanas após o nascimento | Nascimento |
| Posição preferida | Comum | Rara |
| Torcicolo | Presente | Ausente/Presente |
| Crista óssea ao longo da sutura lambdoide | Ausente | Presente |
| Abaulamento mastoide | Ausente | Presente |
| Bossa frontal | Ipsilateral | Contralateral |
| Deslocamento da orelha ipsilateral | Anterior | Posterior |
| Forma do crânio | Paralelogramo | Trapezoidal |
| Diagnóstico | Clínico, por meio de anamnese e exame físico | Tomografia computadorizada tridimensional |
| Tratamento | Clínico | Cirúrgico |


Os pediatras devem fazer o exame físico com a ajuda dos pais; inicialmente, o paciente permanece no colo da mãe/pai de frente para o pediatra e de costas para os pais; finalmente, o examinador deve observar a criança a partir de uma vista superior. Durante o exame físico, as simetrias entre o crânio, a testa e as orelhas devem ser cuidadosamente analisadas.
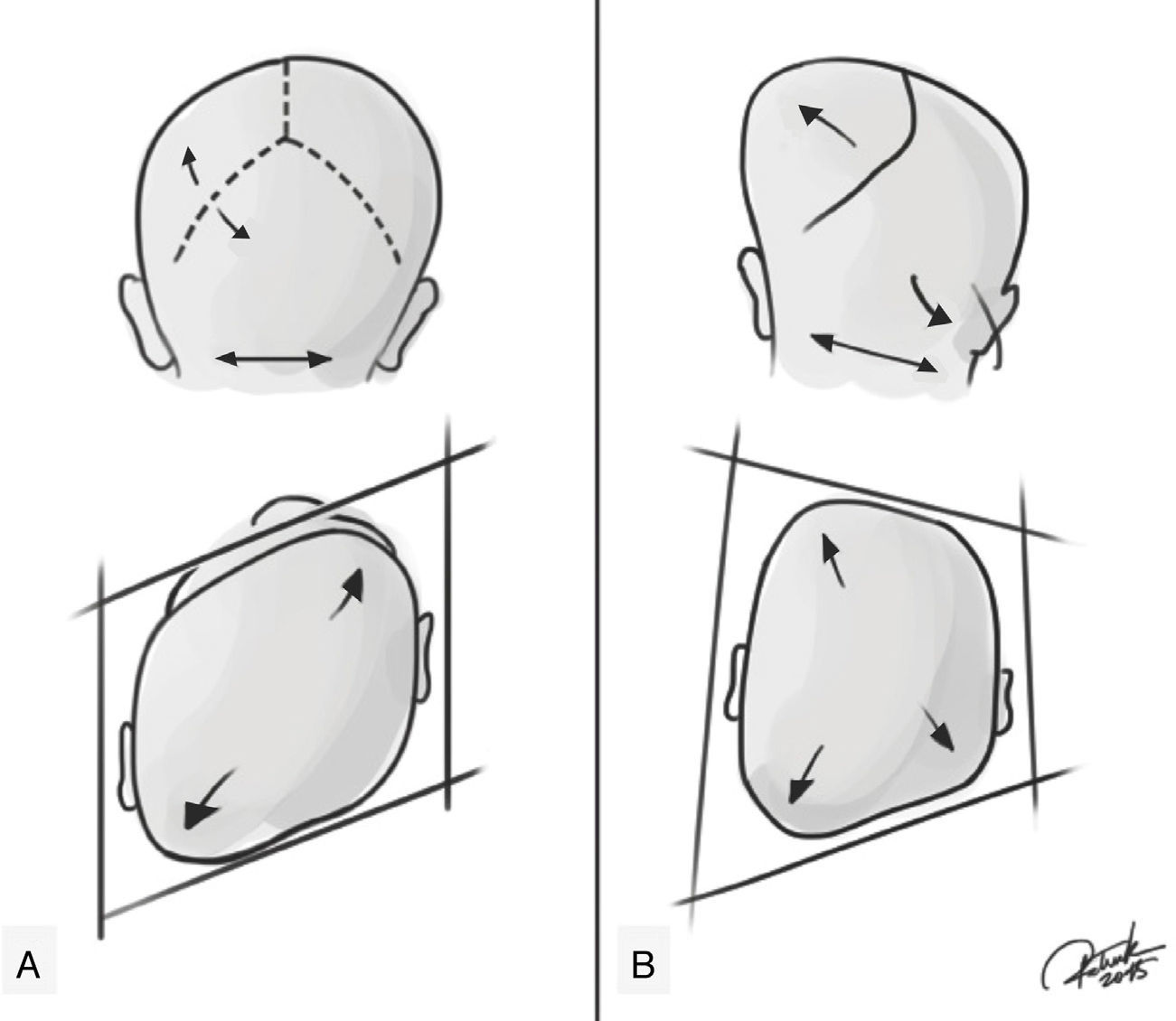
A plagiocefalia posicional apresenta um crânio em formato de paralelogramo, enquanto na plagiocefalia sinostótica posterior o formato é trapezoide.32 Adicionalmente, um abaulamento ipsilateral na região mastoidea e uma saliência na sutura lambdoide fundida podem ser observados e palpados. Em casos moderados a graves em ambas as deformidades, uma bossa frontal compensatória pode ser observada, ipsilateral em plagiocefalia posicional e contralateral em plagiocefalia sinostótica. Esse acometimento da testa pode progredir, levar a uma escoliose facial em ambas as patologias e alterar a simetria facial. Em pacientes com craniosinostose de sutura lambdoide, a estenose da orelha ipsilateral tende a estar em uma posição posterior e para baixo, como se a sutura a puxasse. Enquanto a plagiocefalia posicional tende a estar em uma posição anterior, como se tivesse sido empurrada (fig. 2). O exame físico também deve incluir a avaliação da região cervical e procurar evidências de torcicolo congênito e/ou espessamento do músculo esternocleidomastoideo, que estão diretamente relacionados com a plagiocefalia posicional.
. (A) Plagiocefalia posicional que mostrando ausência de estenose de sutura lambdoide, crânio em formato de paralelogramo, bossa frontal compensatória ipsilateral, orelha ipsilateral em uma posição anterior, como se tivesse sido empurrada. (B) Plagiocefalia posterior verdadeira que mostra presença de estenose de sutura lambdoide, forma trapezoidal, abaulamento ipsilateral na região mastoide, bossa frontal compensatória contralateral; a estenose da orelha ipsilateral tende a estar em uma posição posterior e para baixo, como se a sutura a puxasse. Créditos: Patrick Braga.")
Representação de plagiocefalia posicional e plagiocefalia posterior verdadeira (sinostótica). (A) Plagiocefalia posicional que mostrando ausência de estenose de sutura lambdoide, crânio em formato de paralelogramo, bossa frontal compensatória ipsilateral, orelha ipsilateral em uma posição anterior, como se tivesse sido empurrada. (B) Plagiocefalia posterior verdadeira que mostra presença de estenose de sutura lambdoide, forma trapezoidal, abaulamento ipsilateral na região mastoide, bossa frontal compensatória contralateral; a estenose da orelha ipsilateral tende a estar em uma posição posterior e para baixo, como se a sutura a puxasse. Créditos: Patrick Braga.
O diagnóstico e o tratamento de plagiocefalia posicional são clínicos.32 Algumas orientações devem ser transmitidas aos pais na primeira consulta de puericultura, como evitar posições de má postura para dormir ou no trocador, notar a presença de torcicolo, deixar a criança o menor tempo possível no bebê‐conforto e incentivar o bebê a ficar mais tempo em decúbito ventral, enquanto sob supervisão. É importante diagnosticar qualquer restrição cervical (p. ex., torcicolo congênito ou espessamento do músculo esternocleidomastoideo) e orientar os pais sobre a necessidade de tratamento fisioterápico precoce.
Além das orientações, medidas terapêuticas podem ser empregadas, como forma de forçar o bebê a dormir sobre o lado contralateral da deformidade, incentivar a mudança da localização do berço e do trocador para forçar o bebê a virar a cabeça para o lado em que ele está, além de estimular o bebê a sentar‐se. Tais medidas são eficazes até os 4‐6 meses como tratamento da plagiocefalia posicional.32,33
A prevalência de deformidade posicional tende a diminuir com a idade e pode ser tão baixa como 3,3% aos dois anos, o que evidencia a capacidade natural de remodelação do crânio.32 Contudo, após os seis meses de idade, o uso de um capacete específico pode ser indicado em pacientes com deformidades graves para ajudar na remodelação do crânio. Devemos ressaltar que o capacete requer pelo menos 23 horas de uso por dia para ser eficaz, o que pode resultar em úlceras de pressão e abrasões locais, além do elevado custo do aparelho e do incômodo para a criança.34
ConclusõesAs deformidades cranianas são queixas comuns e altamente prevalentes na rotina do pediatra. Embora a grande maioria das crianças apresente deformidades posicionais, o diagnóstico precoce de craniossinostose e o encaminhamento para tratamento especializado em tempo hábil é fundamental para aprimorar os resultados cirúrgicos. O diagnóstico das deformidades posicionais é geralmente clínico e o tratamento consiste em orientações e medidas para prevenir o agravamento da condição simples. Diante do diagnóstico de craniossinostose, uma abordagem multidisciplinar da criança com craniossinostose é crucial para uma taxa de sucesso cirúrgico maior e para minimizar as complicações.
FinanciamentoO estudo não recebeu financiamento.
Conflitos de interesseOs autores declaram não haver conflitos de interesse.
