


Descrever o caso de um lactente portador de doença de Hirschsprung na forma aganglionose colônica total que, após a ressecção cirúrgica do segmento agangliônico, manteve quadro irreversível de obstrução intestinal funcional; discutir as dificuldades no manejo dessa forma da aganglionose congênita e apontar um mecanismo patogenético plausível para o caso.
Descrição do casoO diagnóstico de doença de Hirschsprung na forma aganglionose colônica total foi definido em lactente aos dois meses de vida, após episódio de enterocolite, choque hipovolêmico e desnutrição grave. Após ressecção colônica, o paciente não recuperou a função motora intestinal que possibilitasse a alimentação via enteral. O exame do íleo remanescente pós‐operatório mostrou presença de plexos ganglionares e redução numérica das células intersticiais de Cajal em segmentos proximais do intestino. Aos 12 meses de vida, o paciente mantém‐se dependente de nutrição parenteral total.
ComentáriosA doença de Hirschsprung na forma aganglionose colônica total tem particularidades clínico‐cirúrgicas que a diferenciam das formas clássicas e dificultam o diagnóstico e o manejo clínico‐cirúrgico. A evolução pós‐operatória pode associar‐se à morbidade permanente decorrente de dismotilidade intestinal. A redução numérica ou as alterações das conexões neurais das células intersticiais de Cajal podem representar uma possível base fisiopatológica para a condição.
To describe the case of an infant with Hirschsprung's disease presenting as total colonic aganglionosis, which, after surgical resection of the aganglionic segment persisted with irreversible functional intestinal obstruction; discuss the difficulties in managing this form of congenital aganglionosis and discuss a plausible pathogenetic mechanism for this case.
Case descriptionThe diagnosis of Hirschsprung's disease presenting as total colonic aganglionosis was established in a two‐month‐old infant, after an episode of enterocolitis, hypovolemic shock and severe malnutrition. After colonic resection, the patient did not recover intestinal motor function that would allow enteral feeding. Postoperative examination of remnant ileum showed the presence of ganglionic plexus and a reduced number of interstitial cells of Cajal in the proximal bowel segments. At 12 months, the patient remains dependent on total parenteral nutrition.
CommentsHirschsprung's disease presenting as total colonic aganglionosis has clinical and surgical characteristics that differentiate it from the classic forms, complicating the diagnosis and the clinical and surgical management. The postoperative course may be associated with permanent morbidity due to intestinal dysmotility. The numerical reduction or alteration of neural connections in the interstitial cells of Cajal may represent a possible physiopathological basis for the condition.
A doença de Hirschsprung (DH) é a causa mais prevalente de obstrução intestinal funcional em crianças, incide em 1:5.000 nascidos vivos.1 Tem determinação genética e caracteriza‐se por um defeito embrionário na migração de células a partir da crista neural, gerando um segmento agangliônico na extremidade distal do intestino.2 O método padrão‐ouro para o diagnóstico é a biopsia retal, que mostra ausência de células ganglionares e número aumentado de fibras nervosas acetilcolinesterase positivas.3
O local da transição anatômica entre o segmento agangliônico distal e o segmento ganglionar proximal permite classificar a DH nas formas: clássica – quando o segmento agangliônico estende‐se até o sigmoide proximal; com segmento longo – quando a aganglionose alcança a flexura esplênica ou o cólon transverso e a aganglionose colônica total (DHACT) – quando o segmento agangliônico estende‐se desde o ânus até, no máximo, 50cm proximais à válvula ileocecal. A DHACT apresenta diferenças clínicas, histológicas e genéticas em relação às demais e associa‐se a dificuldades diagnósticas e de manejo.4 A forma clássica incide em 7%‐88,8% dos casos, a forma longa em 3,9%‐23,7% e a DHACT em até 12,6% dos pacientes.5
A terapia cirúrgica na DH minimiza as complicações da obstrução intestinal quando o segmento aganglionar é completamente ressecado. Em alguns pacientes, há persistência da dismotilidade intestinal pós‐cirúrgica, mais frequentemente manifestada por constipação crônica e episódios recorrentes de enterocolite. Diferentes achados histopatológicos podem ser identificados nesses casos, como a incompleta ressecção do segmento agangliônico, a hipoganglionose e a displasia intestinal neuronal justaposta à zona aganglionar.4
Apresentamos o relato de um caso em que dificuldades de diagnóstico, de terapêutica e de prognóstico foram observadas. A publicação deste relato de caso foi aprovada pelo Comitê de Ética em Pesquisa da Pró‐Reitoria de Pesquisa da Universidade Estadual de Campinas, Parecer CEP/Artigo n° 012/2015 em 28/07/2015.
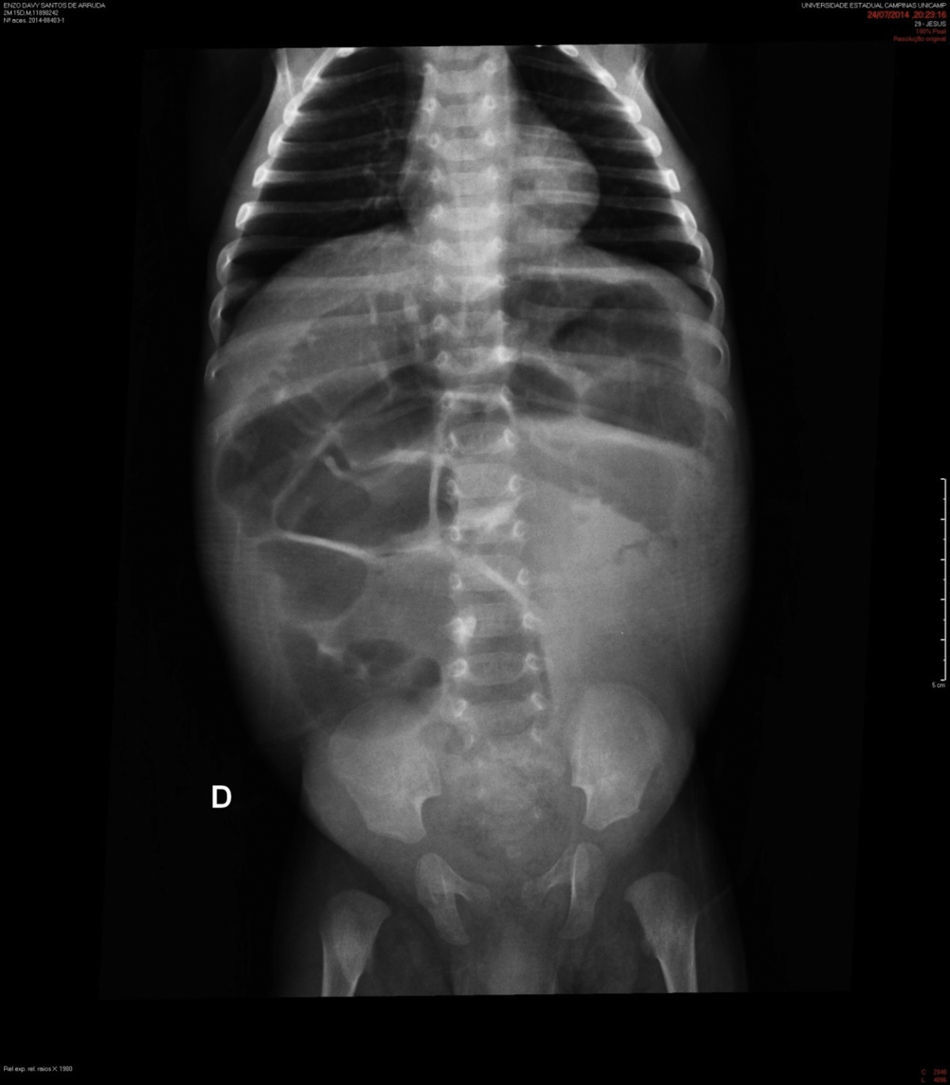
Descrição do casoPaciente masculino, negro, encaminhado ao hospital terciário aos dois meses de vida com diagnóstico de diarreia intratável e vômitos havia 22 dias. Evoluiu com choque hipovolêmico e acidose metabólica refratária. Trazia radiografia simples de abdome que mostrava distensão generalizada de alças intestinais (fig. 1) e tomografia computadorizada que indicava ausência de progressão do contraste enteral para segmentos colônicos distais.

Histórico de ultrassonografia pré‐natal demonstrando distensão de alças intestinais fetais. Mãe negava antecedente de retardo (>24 horas) na eliminação de mecônio ao nascimento e queixas compatíveis com obstrução intestinal. Nascido de parto normal, 39 semanas de idade gestacional, alta hospitalar no terceiro dia de vida, peso ao nascer de 2.950g. Triagem neonatal negativa para fenilcetonúria, hipotireoidismo e fibrose cística. Aleitamento materno exclusivo, desde o nascimento, negava constipação, mas referia baixo ganho ponderal. Aos 38 dias de vida apresentou diarreia que exigiu internação na cidade de origem. Após 10 dias, foi transferido para hospital terciário por diarreia e acidose metabólica intratáveis.
À admissão no hospital terciário, peso 2.860g, atrofia muscular generalizada, tecido subcutâneo escasso, abdome distendido, timpânico à percussão, sem visceromegalias. Orifício anal de implantação tópica e, ao toque retal, esfíncter anal normotônico, ampola retal com dimensões apropriadas. Apresentou eliminação explosiva de grande quantidade de fezes líquidas ao toque. Considerada hipótese diagnóstica de doença de Hirschsprung, foi feita manometria anorretal. No estudo não foi possível registrar reflexo inibitório retoanal. Feita então laparotomia exploradora, visualizou‐se zona de transição 10cm abaixo da válvula ileocecal. Procedida a colectomia total, incluindo 2cm de íleo, e confeccionada ileostomia. Estudo histopatológico do segmento ressecado mostrou ausência completa de corpos neuronais em segmentos colônicos e raros corpos neuronais nos plexos mioentéricos do segmento ileal proximal fixado ao estoma.
No pós‐operatório evoluiu com intolerância (distensão abdominal e vômitos biliosos) a mínimos volumes de fórmula elementar infundidos via sonda nasogástrica. Feita segunda abordagem cirúrgica em que foi descartada a presença de brida ou acotovelamento de alça, foram ressecados 10cm de íleo terminal, foi reconfeccionada a fixação da alça ao estoma e foram feitas biópsias de íleo terminal até a transição com jejuno. Nessas amostras foram identificadas células ganglionares com morfologia e número adequados nos plexos ganglionares. Adicionalmente, as amostras foram fixadas em formalina por 48 horas, desidratadas em álcool e embebidas em parafina. Secções histológicas de 4μm de espessura foram coradas para hematoxilina‐eosina e para imunoistoquímica para células intersticiais de Cajal (CIC). Após aquecimento e incubação com peroxidase, as secções foram incubadas com anticorpo anti‐CD 117, desparafinizadas, colocadas em contato com os anticorpos específicos e coradas. Para controle interno, kits de coloração imunoistoquímica para mastócitos em neurofibroma humano foram usados. CIC foram contadas em dez campos microscópicos de maior aumento (400×) nas proximidades dos plexos mioentéricos. O número médio de células intersticiais de Cajal nos espécimes cirúrgicos variou amplamente entre as secções visualizadas, não guardou relação com a altura em relação à ileostomia (mediana de 1,2 e variação de 0‐9,7 células/CMA). Os valores de cada porção não mostraram diferença quando comparados pelo teste de Mann‐Whitney (p=0,23), mas quando comparados com valores observados em estudos de adultos saudáveis, tais valores estão reduzidos.6
Paciente persiste com necessidade de nutrição parenteral até a presente data, aos 12 meses de vida.
DiscussãoEm até 90% dos pacientes, a DH manifesta‐se no período neonatal, caracteriza‐se por retardo na primeira eliminação de mecônio e distensão abdominal progressiva;4 vômitos biliosos ocorrem em 19%‐37% dos casos.7 O atraso na eliminação do mecônio nas primeiras 24 horas de vida é o indicativo mais forte da condição e é relatado em até 90% dos pacientes com a doença.7 Entretanto, há relatos de que até 40% dos pacientes podem eliminar mecônio nas primeiras horas de vida. Isso revela que esse não é um sinal obrigatório para o diagnóstico.8
As características clínicas da DHACT, em geral, diferem das formas clássicas, a saber, proporção de incidência entre gêneros que nas formas clássicas é de quatro homens para uma mulher, na DHACT é de 1:1. A apresentação clínica com obstrução intestinal neonatal pode não fazer parte do cortejo na DHACT. Não é incomum, nessa forma, o início tardio dos sintomas, nas primeiras semanas de vida ou mais tardiamente ainda, há casos diagnosticados na adolescência.9 Na DHACT, espera‐se a visualização de microcólon no enema baritado, contudo, por vezes, as alças colônicas apresentam calibre normal, sem qualquer indício de obstrução.10 Cerca de 20% dos pacientes com DHACT fazem uma segunda ressecção distal com reposicionamento cirúrgico da alça distal no estoma, visto que a zona de transição é irregularmente disposta e difícil de ser estabelecida no intraoperatório;3 finalmente, 6,5% dos pacientes farão conversão para ileostomia permanente por persistência de disfunção motora.5 No caso em relato, a distensão de alças visualizada na ultrassonografia gestacional poderia ter antecipado o diagnóstico de obstrução intestinal que, adicionalmente, se manifestou menos intensamente nos primeiros dias de vida, talvez por efeito protetor do aleitamento materno exclusivo e sua reconhecida ação procinética e de proteção imunológica.
A ocorrência de diarreia grave num neonato constitui condição de exceção naqueles amamentados exclusivamente ao peito e pode representar evidência de grave complicação da DH ou de outra doença de base. A enterocolite associada à DH é observada em até 50% dos casos, com frequência mais elevada no primeiro trimestre e primeiro ano de vida, pode ocorrer previamente ou após o procedimento cirúrgico.7 A ocorrência pré‐operatória é mais comum quando o diagnóstico é definido após o primeiro mês de vida.7 A complicação manifesta‐se por distensão abdominal, diarreia explosiva, que evoluiu para vômitos, febre, letargia e choque. A diarreia não decorre de uma infecção intestinal específica, mas das características constitucionais do paciente com DH, envolve, possivelmente, uma relação entre a disfunção do sistema nervoso entérico, produção anormal de mucina, secreção insuficiente de imunoglobulinas, desequilíbrio da microbiota intestinal e isquemia da mucosa. A terapia envolve recuperação hidroeletrolítica, descompressão colônica e antibióticos.11
O pós‐operatório na DHACT é caracterizado por alta morbidade, incluindo intolerância alimentar, distúrbios eletrolíticos e desidratação devida à secreção intestinal exacerbada. A consideração antecipada dessas intercorrências diminui a mortalidade, mas, em longo prazo, há maior risco de permanência do estoma por longos períodos nesses pacientes.12 No caso em discussão, a análise do segmento proximal mostrou hipoganglionose, característica da zona de transição, o que pode explicar a necessidade da segunda intervenção.
Técnicas de biologia molecular e anatomia patológica em amostras de pacientes com neuropatias intestinais indicam que agressões inflamatórias e degenerativas decorrentes de isquemia, estase luminal e alterações na microbiota podem comprometer a integridade morfofuncional do sistema nervoso entérico e de outros elementos responsáveis pela motilidade intestinal e resultar em morbimortalidade pós‐operatória.4
Alterações quantitativas ou nas redes de conexões das células intersticiais de Cajal (CIC) têm sido descritas em várias condições que evoluem com dismotilidade gastrointestinal, tais como a acalasia de esôfago, gastrosquise, pseudo‐obstrução intestinal, enterite necrosante, doença inflamatória intestinal e malformação anorretal.13
As CIC desenvolvem‐se a partir do mesoderma embrionário e localizam‐se entre as fibras nervosas e as células da musculatura lisa na parede intestinal. As células são capazes de transmitir impulsos a partir dos neurônios motores entéricos e de gerar atividade elétrica rítmica de onda lenta, coordenam a atividade de marca‐passo e a propagação das ondas lentas, além de atuar na identificação sensitiva de fenômenos de estiramento muscular. No trato gastrintestinal, estão distribuídas desde o esfíncter esofágico superior até o esfíncter interno do ânus,13 justapostas às terminações nervosas dos neurônios motores mioentéricos. A presença do receptor da tirosina quinase na membrana celular permitiu o desenvolvimento de anticorpos anti‐c‐kit para identificação histológica dessas células.14 A ausência ou redução no número das CIC determina anormalidades nas ondas lentas, causa diminuição da contratilidade das células musculares lisas e diminui a velocidade de trânsito do conteúdo intestinal.15
No intestino agangliônico e na zona de transição em DH, pode haver redução numérica e/ou distúrbios nas redes de comunicação das CIC c‐kit positivo.16 Poucos estudos avaliaram a relação entre DH e a distribuição das CIC no segmento remanescente ganglionar. A pesquisa dessas células no segmento intestinal remanescente ganglionar de 15 crianças identificou contagem normal em 13 casos, que evoluíram sem dismotilidade pós‐operatória. Em dois casos, contudo, o estudo histopatológico indicou redução significativa na contagem das células de Cajal e os pacientes evoluíram com constipação grave e persistente, necessitaram de nova ressecção do segmento dilatado remanescente.17 Em outra série de casos, a contagem das CIC no cólon ganglionar de 11 crianças com DH foi comparada com a contagem em grupo controle. Um dos pacientes apresentou número reduzido e evoluiu com constipação grave.18 Gfroerer e Rolle avaliaram os relatos de casos disponíveis e concluíram que existe considerável heterogeneidade na expressão das CIC no intestino ganglionar dos pacientes com DH e que os diferentes padrões de expressão poderiam decorrer de diferenças no tamanho dos segmentos agangliônicos, no tempo até a ressecção do segmento aganglionar e, ainda, nos métodos de contagem das células.19
Esses achados requerem interpretação cautelosa, tendo em vista a escassez de estudos que relacionem o número das CIC e a função motora em humanos. Além disso, as amostras de tecido examinadas são provenientes de pacientes que apresentaram doença obstrutiva durante diferentes períodos, o que torna difícil determinar se a redução é uma consequência do processo de obstrução ou uma associação patogenética à DH. A diversidade dos defeitos das CIC pode indicar que esse fenômeno seja secundário às alterações decorrentes da obstrução intestinal, já que as CIC são particularmente sensíveis à isquemia.14,15 A incapacidade de visualização de todas as células a partir de amostras fixadas em formalina e parafina é um fator adicional que dificulta a definição do diagnóstico histopatológico.19
No paciente em discussão, não foi possível comparar a amostra histológica com a de crianças saudáveis, um material dificilmente disponível.20 Amostras apropriadas de tecido, manuseio correto e perícia em interpretação anatomopatológica são cruciais para aumentar a acurácia do diagnóstico de redução numérica das CIC. Seria interessante que um maior número de pacientes com evolução desfavorável fosse examinado para determinar o significado do achado histopatológico de diminuição das CIC e sua correlação com a clínica.
FinanciamentoO estudo não recebeu financiamento.
Conflitos de interesseOs autores declaram não haver conflitos de interesse.
