


PROP1 (Prophet of POUF1) mutations are the most frequent genetic cause of combined pituitary hormone deficiency, a condition associated with a deficiency or inadequate production of hormones of the anterior pituitary. The PROP1 gene encodes a transcription factor involved in the ontogeny, differentiation and function of somatotrophs, lactotrophs and thyrotrophs. These mutations are characterized by a remarkable clinical variability, including time of onset of hormonal deficiencies, hypophyseal dimensions and secretion of cortisol.
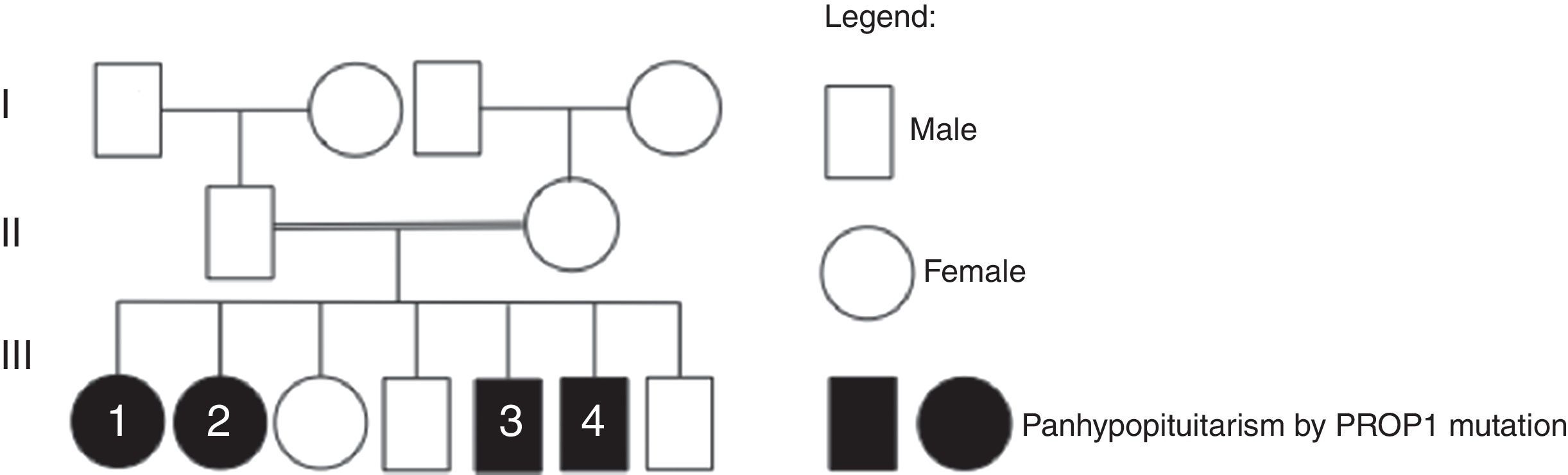
Case reportWe describe a family of consanguineous parents (second-degree cousins), composed of 7 siblings, 4 with combined pituitary hormone deficiency. Two brothers, 41 and 45 years of age, had an initial diagnosis of dwarfism at ages 9 and 12 respectively. Subsequently, TSH, FSH/LH and prolactin deficiency was detected in both. The latter was also diagnosed with cortisol deficiency. The two sisters, aged 46 and 50-years-old, were diagnosed with combined pituitary hormone deficiency, namely of GH, TSH, FSH/LH, prolactin and ACTH, since the ages of 15 and 9, respectively. There was no previous family history of combined pituitary hormone deficiency. The genetic study was performed in the 4 brothers, detecting a homozygous mutation in the PROP1 gene (c.301–302delAG).
ConclusionThis case reflects the variability of clinical expression and the progressive functional impairment, including pituitary secretion of ACTH, in patients with PROP1 gene mutations.
As mutações no gene PROP1 (Prophet of POUF1) são a causa genética mais frequente de insuficiência hormonal combinada, uma condição associada à deficiência ou produção inadequada de hormonas da hipófise anterior. O gene PROP1 codifica um factor de transcrição envolvido na ontogênese, diferenciação e função dos somatotrófos, lactotrófos, e tireotrófos. Estas mutações caracterizam-se por uma notável variabilidade clínica, incluindo o início do aparecimento das deficiências hormonais, dimensões hipofisárias e secreção de cortisol.
Caso clínicoFamília de pais consanguíneos (primos em segundo grau), composta por 7 irmãos, 4 com o diagnóstico de insuficiência hormonal combinada, seguidos em consulta de Endocrinologia. Dois irmãos do sexo masculino, 41 e 45 anos, com diagnóstico inicial de nanismo aos 9 e 12 anos de idade, respetivamente, tendo sido detetada posteriormente deficiência de TSH, FSH/LH e prolactina, em ambos e também de cortisol no último. As 2 irmãs, de 46 e 50 anos de idade, com insuficiência hormonal combinada por deficiência de GH, TSH, FSH/LH, prolactina e ACTH, confirmada aos 15 e aos 9 anos de idade, respetivamente. Sem história familiar prévia de insuficiência hormonal combinada. Foi efectuado o estudo genético, tendo sido possível detectar nos 4 irmãos uma mutação homozigótica no gene PROP1 (c.301–302delAG).
ConclusãoEsta família demonstra a variabilidade da expressão clínica dos portadores de mutações do gene PROP1 e a progressiva alteração funcional hipofisária, nomeadamente da secreção de ACTH.
The anterior pituitary development during the embryonic period is dependent on sequential and critical processes, from induction of Rathke's pouch to pituitary organogenesis and cell line proliferation, specification and differentiation. Deregulation of these steps can cause pituitary hypoplasia or dysfunction. Molecular analysis has allowed the identification of multiple genes involved in the coordination of this differentiation.1 POUF1 gene encodes a transcriptional factor expressed during the differentiation of the anterior pituitary gland, which is responsible for the adequate production of growth hormone (GH), prolactin (PRL) and thyroid-stimulating hormone (TSH). A second transcription factor involved in pituitary development is the gene Prophet of POUF1 (PROP1).2
Mutations in PROP1 gene are the most frequent genetic cause of combined pituitary hormone deficiency, a condition associated with a deficiency or inadequate production of hormones of the anterior pituitary.3 Prop1 gene (Prophet of POUF1, OMIM 601538) is located on chromosome 5q35.31 and encodes a 226-amino acid transcription factor.4 PROP1 mutations have been firstly described in Ames dwarf mice.5 Ames dwarf mutants carry a homozygous mutation that involves a serine-to-proline substitution of amino acid 83 (S83P), associated with multiple hormone deficiencies, namely GH, PRL, TSH and LH and FSH and with adult pituitary hypoplasia. Since then, several Prop1 mutations that structurally affect the ‘paired-like’ DNA-binding domain of the Prop-1 protein molecule have been described.1
Combined pituitary hormone deficiency by PROP1 mutations is an autosomal recessive disorder, and most of the patients do not present any family history of the disease. Thus, each sibling has 25% risk of being affected.6 Since PROP1 gene encodes a transcription factor involved in the ontogeny, differentiation and function of somatotrophs, lactotrophs, gonadotrophs and thyrotrophs, patients with PROP1 mutations show combined pituitary hormone deficiency.7 They are characterized by a remarkable clinical variability, including time of onset of hormonal deficiencies, hypophyseal dimensions and secretion of cortisol. In a multicentric study launched in Portugal to screen for mutations of the PROP1 gene in patients with combined pituitary hormone deficiency, the most frequent mutation found was c.301–302delAG, also known as 296delGA. This 2-bp deletion in a dinucleotide repeat mutational hotspot is also the most prevalent mutation described in other studies.6
Case reportWe describe a family of consanguineous parents (second-degree cousins), composed of 7 siblings, of which 4 present combined pituitary hormone deficiency and have been followed by the department of Endocrinology (family heredogram in Fig. 1). There was no previous family history of combined pituitary deficiency.

Patient #1, the older sister, aged 50-years-old, had unremarkable neonatal period and psychomotor development. Failure to thrive was noticed when she was 9-years-old. Afterwards, documentation of GH, TSH, FSH/LH, prolactin and cortisol deficiency confirmed the diagnosis of combined pituitary hormone deficiency. She had initiated growth hormone therapeutic and, at adult age, she presented a final stature of 1.47m and a body mass index of 32.9kg/m2. She is currently being supplemented with levotiroxine 250mcgqd, hydrocortisone 25mgqd and desogestrel/ethynil estradiol (0.15/0.02mg).
Patient #2, female, aged 46, was also born by normal delivery, at term, and presented an unremarkable neonatal and psychomotor development. Growth retardation and lack of pubertal development were noticed when she was 15-years old. Documentation of GH, TSH, FSH/LH, prolactin and ACTH deficiency confirmed the diagnosis of combined pituitary hormone deficiency. She was treated with adequate complete hormone replacement therapy, acquiring an adult stature of 1.5m (BMI of 35.97m2). She is currently under levotiroxine 250mcgqd, hydrocortisone 20mgqd and estradiol valerate/Norgestrel (2/0.5mg).
Patient #3, male, 45-years-old, also did not show any complication during neonatal period or concerning psychomotor development. An initial diagnosis of dwarfism was performed at age 12. Subsequently, it was detected TSH, FSH/LH and prolactin deficiency, as well as cortisol deficiency. With adequate hormonal supplementation, including growth hormone therapeutic, at adult age he reached 1.68m of stature (BMI of 30.67kg/m2). He is medicated with levothyroxine 200mcgqd, hydrocortisone 25mgqd and testosterone decanoate 4/4 weeks.
Patient #4, male, the youngest sibling, 41-years-old, was born by normal delivery, at term, with a normal birth length. His neuropsychomotor development was normal. Growth retardation was the first signal at 9-years old. Clinical data and standard stimulation tests allowed the detection of TSH, FSH/LH and prolactin deficiency, leading to the diagnosis of combined pituitary hormone deficiency. He did hormone replacement therapy, including growth hormone treatment, acquiring a final stature at adult stage of 1.51m (BMI of 26.18kg/m2). Up until now, he maintains adrenal sufficiency and is presently being supplemented with levothyroxine 225mcg and testosterone decanoate 4/4 weeks.
Pituitary morphologyPituitary morphology by imaging study was only performed in patients #1, 3 and 4.
Patients #3 and #4 had performed a CT scan, which revealed no alterations. On patient #1, MRI showed pituitary hypoplasia.
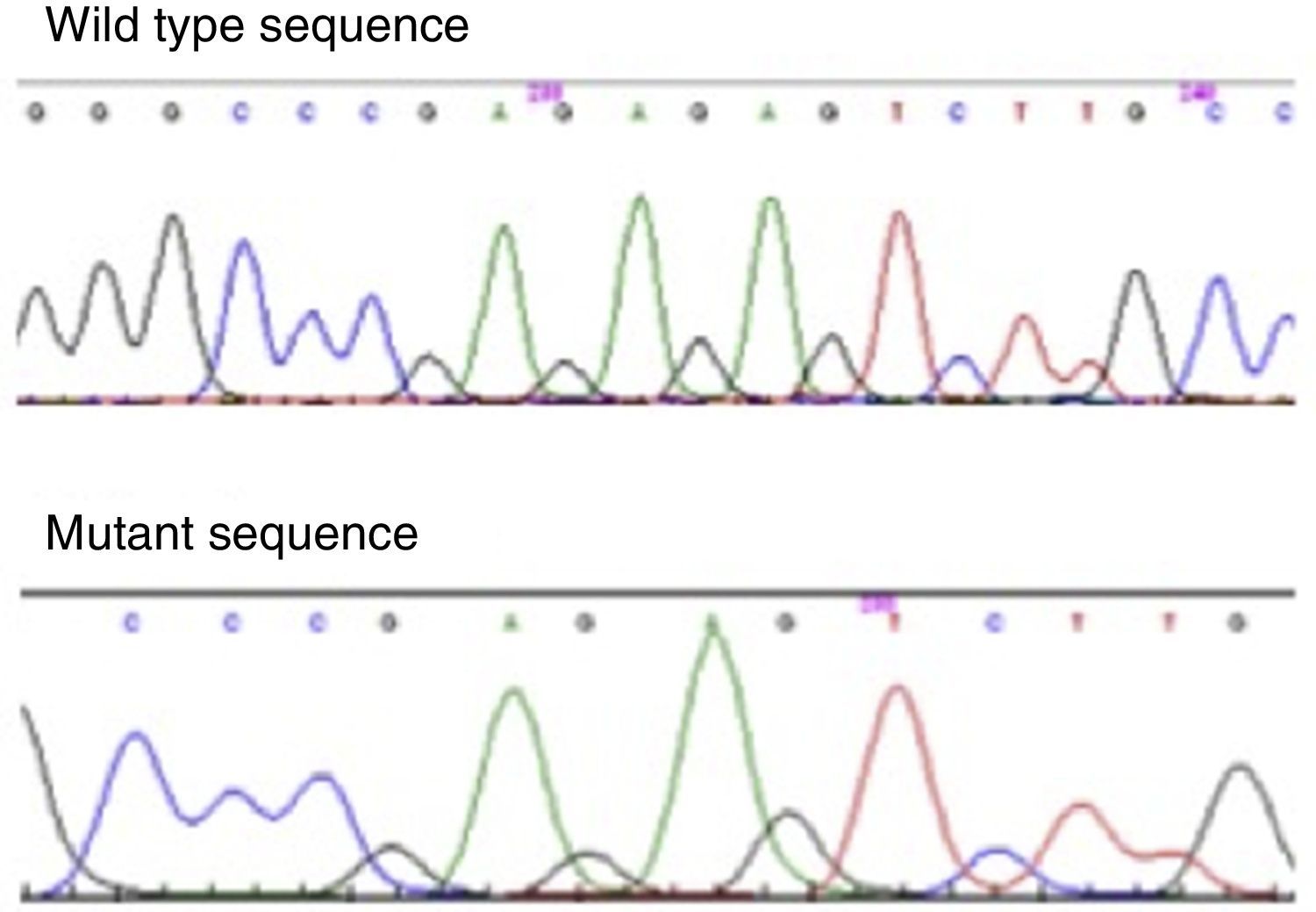
Molecular genetic analysisThe genetic study was performed in the 4 brothers, amplifying PROP1 gene by polymerase chain reaction. It has allowed the identification of a homozygous mutation in the PROP1 gene with a 2-bp deletion (c.301–302delAG). This mutation involves a 2bp GA or AG deletion among three tandems GA repeats (296-GAGAGAG-302) of exon II (Fig. 2). The change in reading frame causes a divergence in amino acid sequence at codon 102, and the serine at codon 109 is changed to a stop codon (S109X). The resulted truncated gene is inactive, which leads to the described hormone abrogation.
Discussion
Pituitary development depends on a high complex process, involving a sequential expression of transcription factors, responsible in the end for an adequate production of the five types of cell populations – somatotrophs, lactotrophs, thyrotrophs, gonadotrophs, and corticotrophs.8 Thereby, any disturbances of these transcriptional factors can have deleterious effects on pituitary development and hormonal production. Combined pituitary hormonal deficiency genetically determined is due to mutations in the PROP1 gene in nearly half of the cases.3
PROP1 expression is essential to allow Pit1-specific cells and the gonadotropic lineage differentiation. PROP1 mutations are characterized by a notably high variable phenotype presentation. GH and TSH deficiency are frequently the first manifestations of disease. Usually, the first signs, namely progressive growth failure, only occur in childhood, not before 6 years of age. Perinatal signs of hypopituitarism are rare, comparing to other genetic forms of combined hormonal pituitary deficiency.9 Lactotroph function and hypogonadism are also different among patients with PROP1 mutation. Prolactin production differs between patients, although usually they are at lower levels than normal.10 Concerning puberty development, its onset depends on the patient; some do not produce sufficient LH and FSH to initiate puberty, whereas others develop progressive gonadotroph failure and do not complete puberty.11 These observations lead to the hypothesis that PROP1 gene may play a key role in gonadotroph differentiation.12 Other authors, taking into account experimental observations, consider that PROP1 directly activates gonadotropin gene expression.13 In our case report, the first sign of disease was growth retardation and the two brothers were firstly diagnosed of dwarfism at the age of 12 and 9, being the other hormonal deficiencies (TSH, prolactin and FSH/LH) diagnosed later.
Adrenal insufficiency seems to be controversial, although there is growing evidence that ACTH impairment is a late-onset complication in patients with PROP1 mutations.14 In a retrospective longitudinal analysis of nine patients with PROP1 mutations, all patients developed at least partial cortisol insufficiency and a progressive decline of the pituitary–adrenal axis was observed.9 Therefore, anterior pituitary function seems to be a continuum that tends to deteriorate progressively and adrenal insufficiency is a perfect example of this condition. From this perspective, PROP1 may also play a role in corticotroph cells differentiation and it is possible that these cells suffer progressive apoptosis with subsequently decrease of ACTH secretion.15 The inexistence of adrenal insufficiency at one point does not exclude the development of inadequate production of cortisol later. As we observed, all except one of our patients affected by PROP1 gene mutation developed adrenal insufficiency, being essential to monitor the adrenal function of the patient that has not yet been affected.
As we might conclude, the progressive disruption of all anterior pituitary axis in patients carrying PROP1 mutations has important clinical consequences regarding diagnosis, prognosis and management of the patients.16 Clinicians should be aware of these defects, monitoring hormonal status since the diagnosis, even if patients do not present all hormonal deficiencies at the initial stages, as they are at increased risk of developing them. Unfortunately, it has been impossible to predict the time point of occurrence of these changes up until now, which implies a strict and careful follow-up.
Pituitary size or morphologic alterations are also matter of debate. While some patients present normal pituitary gland on imaging studies, others have hypoplasic pituitary gland.17 One Portuguese study conducted in patients with PROP1 mutations found out that all patients had pituitary hypoplasia or empty sella except one, who presented an enlarged sellar mass.6 One explanation for this is that this enlargement, generally presented in early childhood, will be followed by regression and involution of that tissue, resulting in an empty sella.18 Out of the 3 patients of our case report that were evaluated by imaging study, two had normal pituitary gland and the last one had pituitary hypoplasia.
It is also difficult to establish a concrete correlation between genotype–phenotype among the diverse forms of presentation of this disease and the different degree of loss function between the reported mutations.19 As we observed in this family, there is a strong variability either at the age of diagnosis, the time of onset of hormone deficiencies, pituitary morphology and ACTH and cortisol status, besides the 4 patients were affected by the same mutation. This observation emphasizes the impact of interindividual differences on clinical phenotype by PROP1 mutations. Furthermore, it seems that these differences tend to be attenuated throughout the years, as hormone insufficiency in adults seems to be similar among the subjects.
ConclusionThis case study exemplifies the clinical expression of combined pituitary hormone deficiency due to PROP1 mutations. As we observed in this family, mutations in PROP1 gene are associated with a variable phenotype expression, from time of onset of hormonal abnormalities, hormonal status, namely cortisol sufficiency, to pituitary morphologic changes. In this regards, an early diagnosis and a careful long-term follow-up are crucial to improve overall patients’ clinical condition, as anterior pituitary function declines gradually.
Ethical disclosuresProtection of human and animal subjectsThe authors declare that no experiments were performed on humans or animals for this investigation.
Confidentiality of dataThe authors declare that no patient data appears in this article.
Right to privacy and informed consentThe authors declare that no patient data appears in this article.
Conflicts of interestThe authors declare no conflicts of interest.
