


Introdução: A Hiperplasia Congénita da Suprarrenal (HCSR) é uma doença autossómica recessiva, cuja gravidade é determinada pela atividade permitida pelo alelo menos afetado. A causa mais comum é a mutação no gene CYP21 que codifica a enzima 21-hidroxilase, sendo a clínica inversamente proporcional à atividade da enzima. As formas de apresentação são: Clássica (perdedora de sal (PS) e virilizante simples (VS)) e Não Clássica. Objetivo: Verificar a existência de correlação genótipo-fenótipo nos doentes com HCSR por défice da 21-hidroxilase.
Métodos: Estudo retrospetivo de 37 pacientes com o diagnóstico de HCSR, definido clínica e analiticamente, seguidos na Consulta Externa de Endocrinologia Pediátrica entre 1999 e 2010. Foi realizada a pesquisa de mutações do CYP21A2 e estabelecida correlação genótipo-fenótipo.
Resultados: Nos 37 pacientes com clínica de HCSR identificaram-se 42 mutações, cinco das quais em heterozigotia não composta, havendo portanto um alelo não mutado. Em 18 crianças (12 do sexo feminino) houve identificação de pelo menos duas mutações no gene CYP21A2, confirmando o diagnóstico de HCSR. Destes, sete apresentavam as duas mutações em homozigotia (39%); um três mutações e outro um alelo mutado associado a uma duplicação do CYP21A2. Nos restantes nove doentes foram identificadas heterozigotias compostas. A apresentação clínica dos casos confirmados por estudo molecular foi: Clássica PS em 27,8% (5), Clássica VS em 38,9% (7) e Não Clássica em 33,3% (6). Verificou-se uma concordância genótipo-fenótipo de 80% nas formas clássicas PS; de 43% nas clássicas VS e de 66,7% nas não clássicas. A concordância total foi de 61,1%.
Conclusões: A elevada proporção de mutações em homozigotia alerta para a possibilidade de consanguinidade nesta amostra. A predominância do sexo feminino com HCSR poderá traduzir uma menor valorização dos sinais de virilização nos rapazes. Atendendo ao alto grau de concordância genótipo-fenótipo e à sua aplicabilidade na prática clínica, os autores reforçam a importância da pesquisa das mutações para confirmação e caracterização da HCSR.
Introduction: Congenital adrenal hyperplasia (CAH) is an autosomal recessive disease. The severity is determined by the activity of the less affected allele. It is most commonly caused by a mutation in the CYP21 gene encoding the 21-hydroxylase enzyme, with clinical manifestations inversely proportional to the activity of the enzyme. Presentations are: classical (salt wasting (SW) and simple virilizing (SV)) and Non-Classical (NC).
Objective: To determine genotype-phenotype correlation in patients with CAH caused by 21-hydroxylase deficiency.
Methods: Retrospective study of 37 patients with the diagnosis of CAH defined clinically and analytically, followed in the Outpatient Clinic of Pediatric Endocrinology between 1999 and 2010. Mutations of CYP21A2 were analyzed and a genotype-phenotype correlation was established.
Results: Forty two mutations were identified in 37 patients with clinical signs of CAH, five of which were not compound heterozygous, so they had a non-mutated allele. In 18 children (12 girls) at least two mutations were identified in the CYP21A2 gene, confirming the diagnosis of CAH. Seven were homozygous (39%), one had three mutations and another one a mutated allele associated to a duplication of CYP21A2. The remaining nine patients were compound heterozygous. Clinical presentation of the confirmed cases was: Classic SW in 27,8% (5), Classical SV in 38,9% (7) and Non-Classical in 33.3% (6). There was a genotype-phenotype correlation of 80% in classical SW, 43% in the classic SV and 66.7% in NC. The overall agreement was 61.1%.
Conclusions: The high proportion of homozygous alerts us to the possibility of consanguinity in this group. The predominance of females with CAH could mean a lower valorization of virilization signs in boys. Given the high genotype-phenotype correlation and its applicability in the clinical practice, the authors reinforce the importance of researching the mutations to confirm and characterize the CAH.
Introdução
A Hiperplasia congénita da suprarrenal constitui um dos principais erros inatos do metabolismo1, por perda ou diminuição da atividade de uma das cinco enzimas esteroidogénicas envolvidas na biosíntese do colesterol (podendo ou não atingir concomitantemente a biosíntese da aldosterona)2. A deficiência da enzima 21-hidroxilase é responsável por cerca de 90 a 95% dos casos1,2.
O gene que codifica a enzima 21-hidroxilase, denominado CYP21A2, localiza-se no braço curto do cromossoma 6, assim como o seu pseudogene CYP21A1P3-5. O facto de existir alta homologia entre os genes CYP21 parece estar na base de emparelhamentos desiguais durante a meiose, conduzindo à possibilidade de crossing-over desiguais, gerando deleções, duplicações, conversões e/ou transferências de sequências deletérias do pseudogene para o gene ativo3.
É uma patologia de transmissão autossómica recessiva, necessitando de dois alelos mutados para ocorrer doença3. Na maioria dos casos (65-75%) os indivíduos são heterozigóticos compostos2, isto é, apresentam 2 mutações distintas, uma em cada alelo. As manifestações clínicas correspondem à atividade enzimática permitida pelo alelo menos afetado2,3, podendo apresentar vários espectros clínicos6,7.
A forma mais grave corresponde à apresentação clássica perdedora de sal (virilização, hipocortisolismo e insuficiência mineralocorticóide no período perinatal). A forma clássica não perdedora de sal apresenta-se com hipocortisolismo e virilização simples também no período perinatal. No entanto, nos rapazes o diagnóstico pode ser mais tardio, por subvalorização dos sinais de virilização precoce5. A forma mais ligeira é a não-clássica, mais subtil e tardia, manifestando-se por pubarca precoce, irregularidades menstruais, hirsutismo e/ou infertilidade1,8,9. No sexo masculino alguns doentes podem permanecer assintomáticos e ser identificados apenas em estudos familiares.
A 17 hidroxi-progesterona (17OHP) é o marcador de diagnóstico da HCSR com maior relevância1, apresentando valores francamente elevados no estado basal na forma clássica (> 2000 ng/dL). Na forma não-clássica a 17OHP pode estar normal ou ligeiramente elevada no estado basal, todavia apresenta uma elevação significativa após a prova de estimulação com ACTH (> 1500 ng/dL)1.
Vários estudos relatam a existência de uma correlação entre as mutações genéticas identificadas (genótipo) e a severidade da apresentação clínica da HCSR (fenótipo)2,5,9. A concordância parece ser mais elevada em mutações mais graves, que condicionam formas clássicas perdedoras de sal (90-95%), comparativamente a formas sem perda de sal (70-75%) ou não clássicas (45-65%)2,8. Este estudo pretende descrever os resultados das mutações do gene CYP21A2 dos doentes com HCSR e analisar a relação entre o genótipo encontrado e o fenótipo.
Métodos
Realizou-se um estudo retrospetivo que incluíu 37 crianças seguidas em Consulta Externa de Endocrinologia Pediátrica no Hospital Pedro Hispano, entre 1999 e 2010, com alterações clínicas e analíticas compatíveis com o diagnóstico de HCSR. Nestas crianças procedeu-se à colheita de sangue periférico com extração de ADN (ácido desoxirribonucleico) para estudo molecular de mutação no gene CYP21A2, realizado em laboratório comercial (Centro de Genética Clínica S.A., Porto). Inicialmente foi efetuada amplificação pela técnica de PCR (polymerase chain reaction) e posterior sequenciação genética do CYP21A2 onde se situam as mutações mais frequentes (P30L no exão 1, I2 splicing no intrão 2, I172N no exão 4, V281L no exão 7, Q318X e R356W no exão 8). Nos casos em que foi apenas identificado um alelo mutado, foi realizada a sequenciação completa e análise por MLPA (Multiplex Ligation-dependent Probe Amplification) do gene CYP21A2 para deteção de uma segunda mutação em heterozigotia composta.
O critério de inclusão no estudo foi apresentar pelo menos duas mutações no gene CYP21A2 (uma em cada alelo), excluindo-se as restantes crianças.
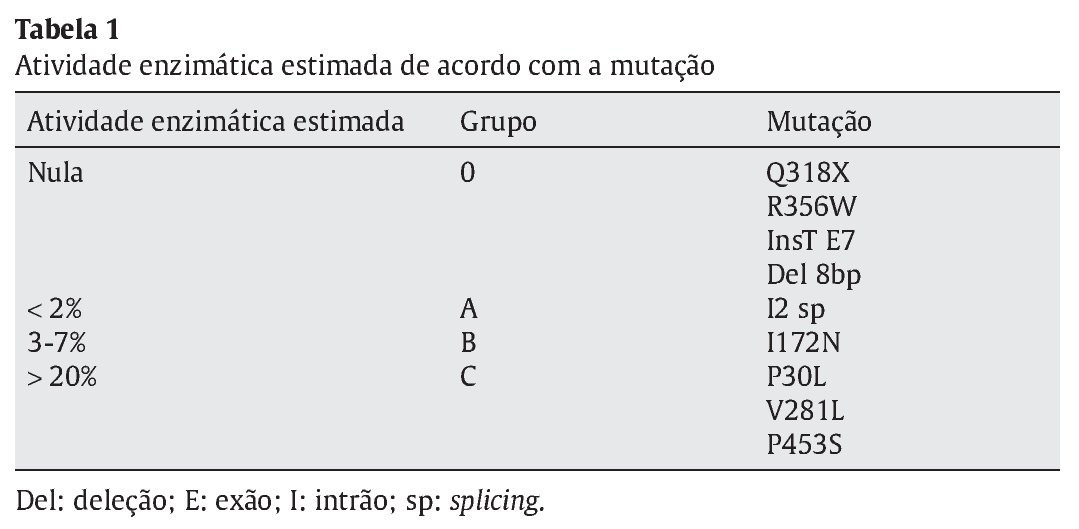
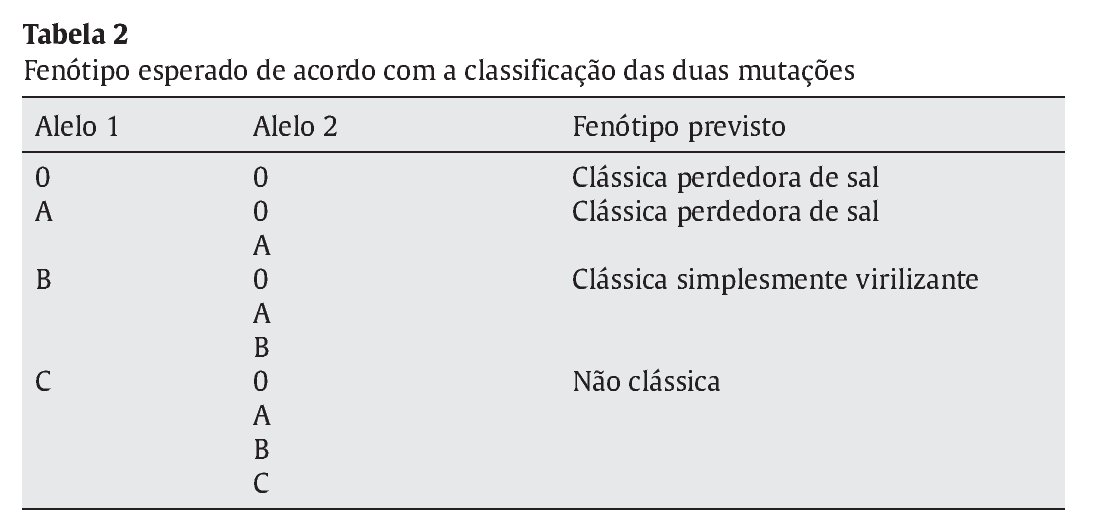
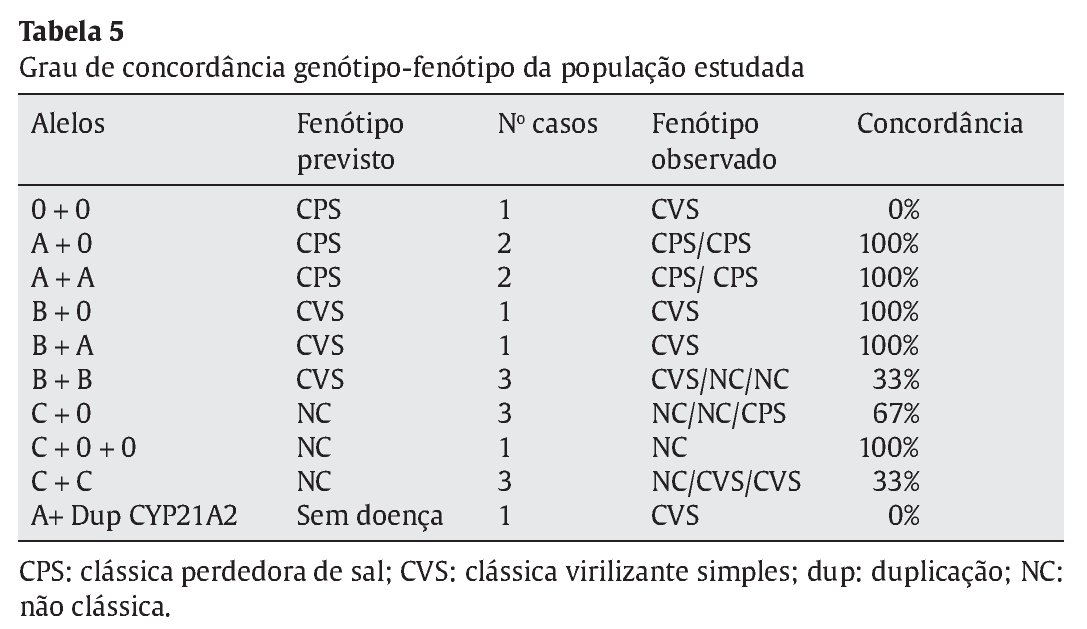
Distribuíram-se as mutações encontradas por 4 grupos conforme o grau de atividade enzimática estimada para cada mutação, de acordo com a literatura- grupo 0 atividade nula, A < 2%; B 3-7% e C > 20%3,8 (Tabela 1). Considerando o conjunto de mutações apresentado por cada doente, estabeleceu-se um fenótipo previsível tendo em conta a atividade enzimática permitida pelo alelo menos afetado (Tabela 2). Efetuou-se a correlação entre o fenótipo previsto e o fenótipo clínico observado e avaliou-se o grau de concordância entre estes.
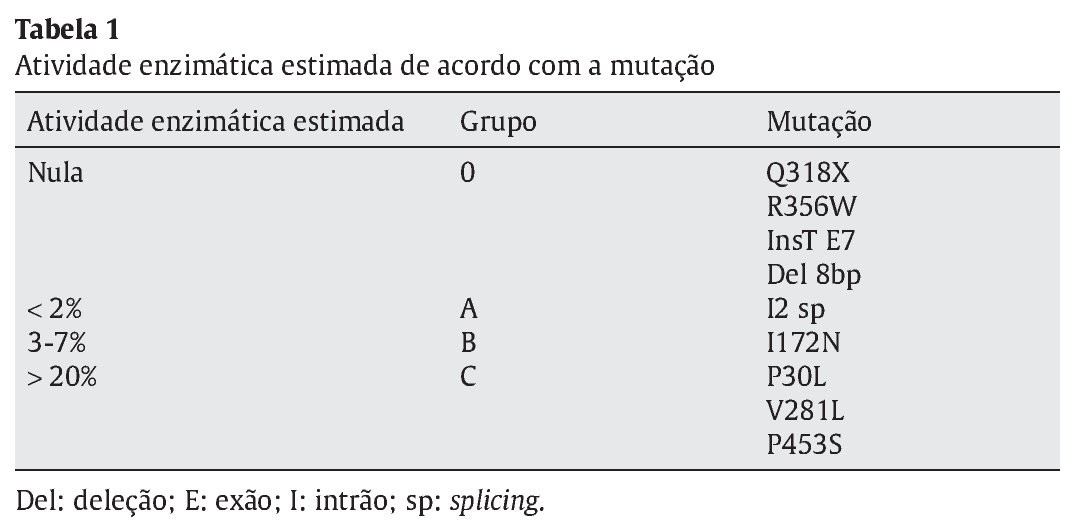
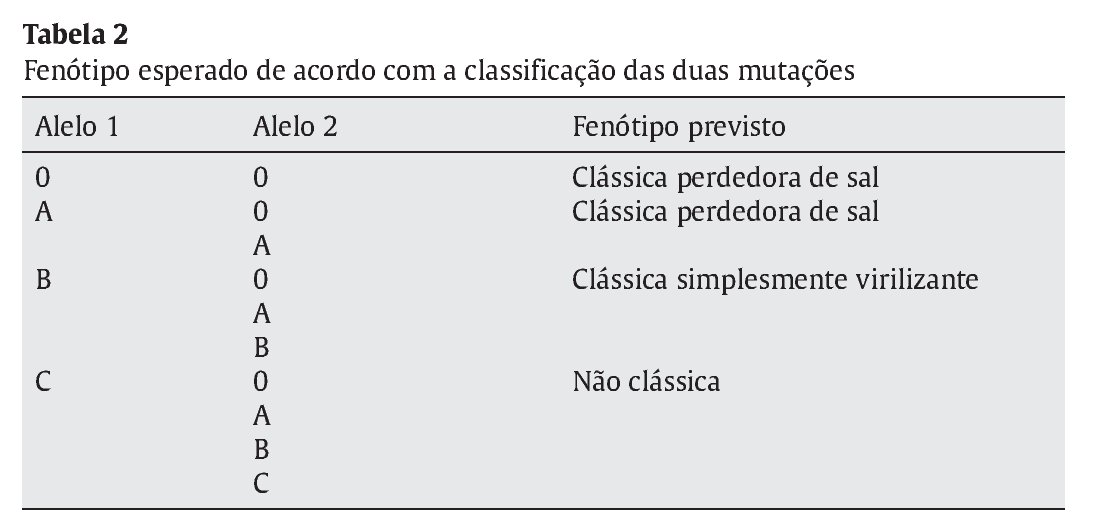
Resultados
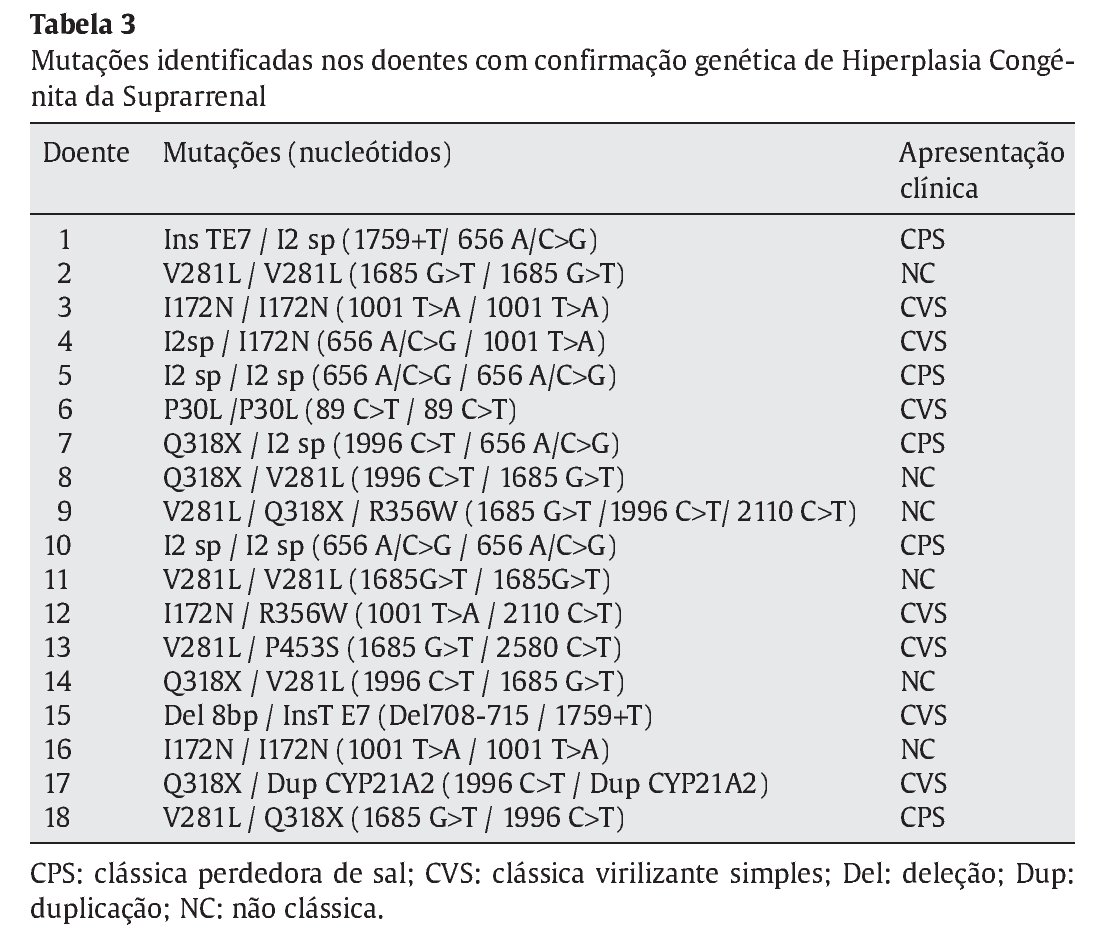
Dos 37 doentes com HCSR, foram identificadas mutações nos dois alelos em 18 crianças (48,6%), confirmando assim o diagnóstico molecular de deficiência de 21-hidroxilase (Tabela 3). Excluíram-se 19 crianças, 14 delas por não se detetar nenhuma mutação e 5 em que se identificou apenas uma. Dos casos em que não foi confirmado o diagnóstico genético, apenas um apresentava manifestações de forma clássica de HCSR sendo os restantes compativeis com uma apresentação não-clássica da doença. Os valores basais de 17-OHP nestes variaram entre 59-2810 ng/dL e nos que apresentavam indicação para realizar prova de estimulação com ACTH a 17-OHP variou entre 287-424 ng/dL.
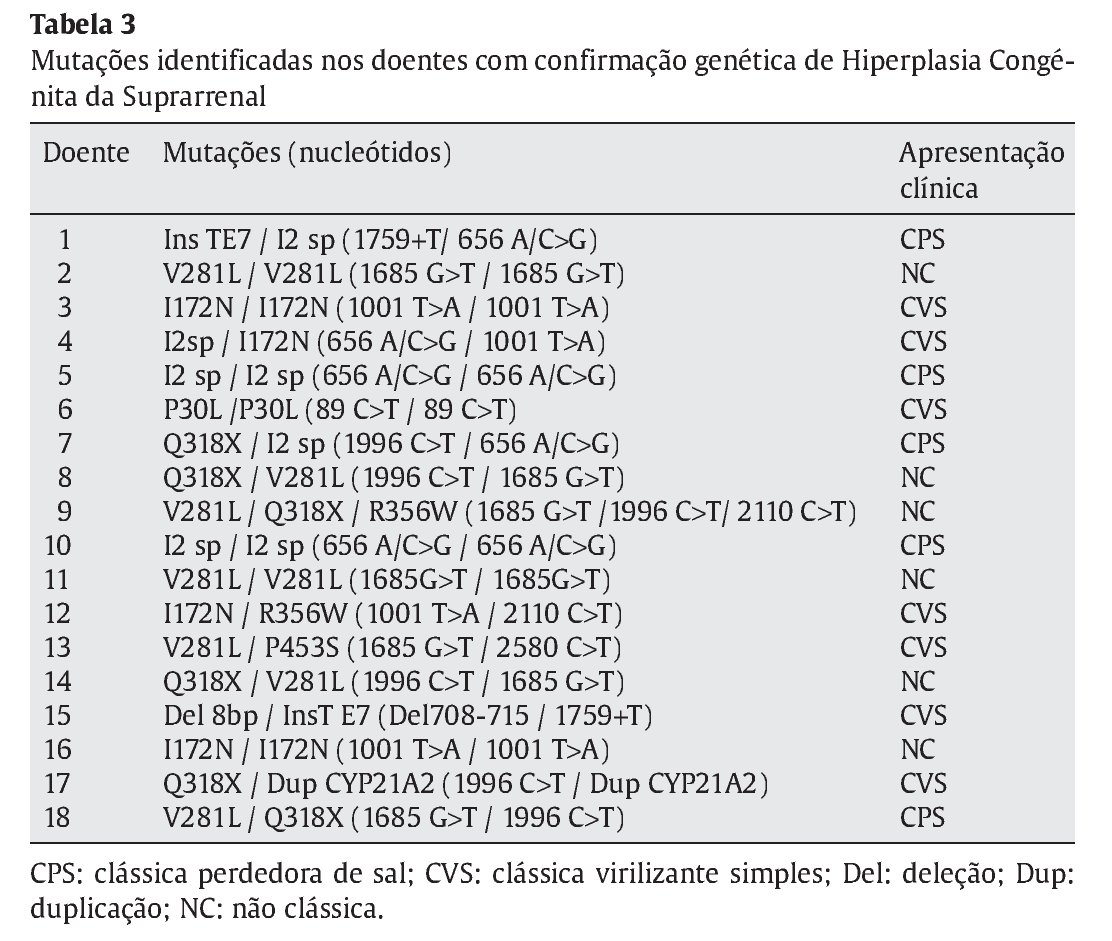
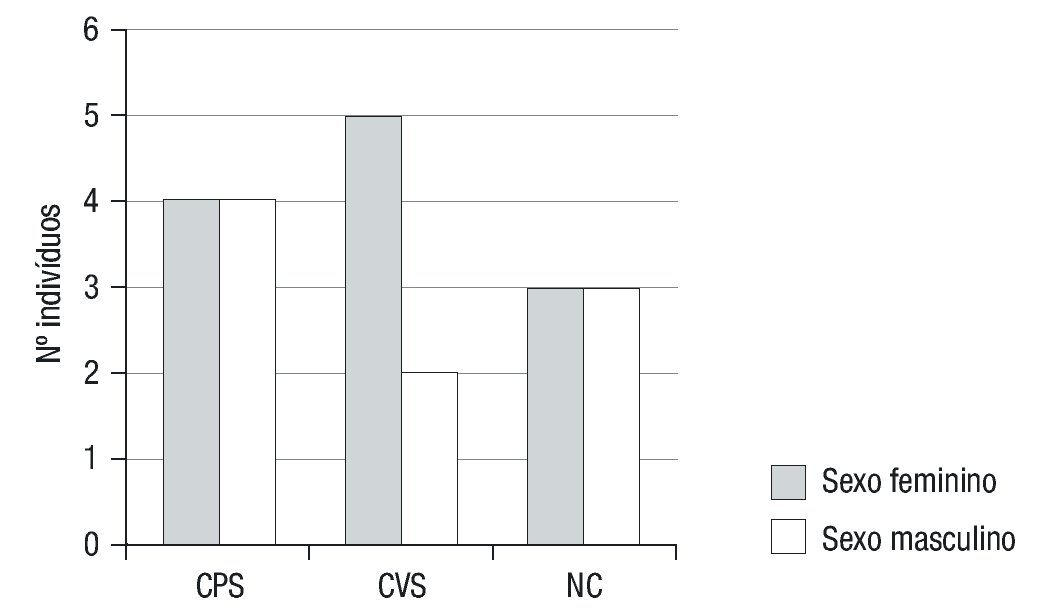
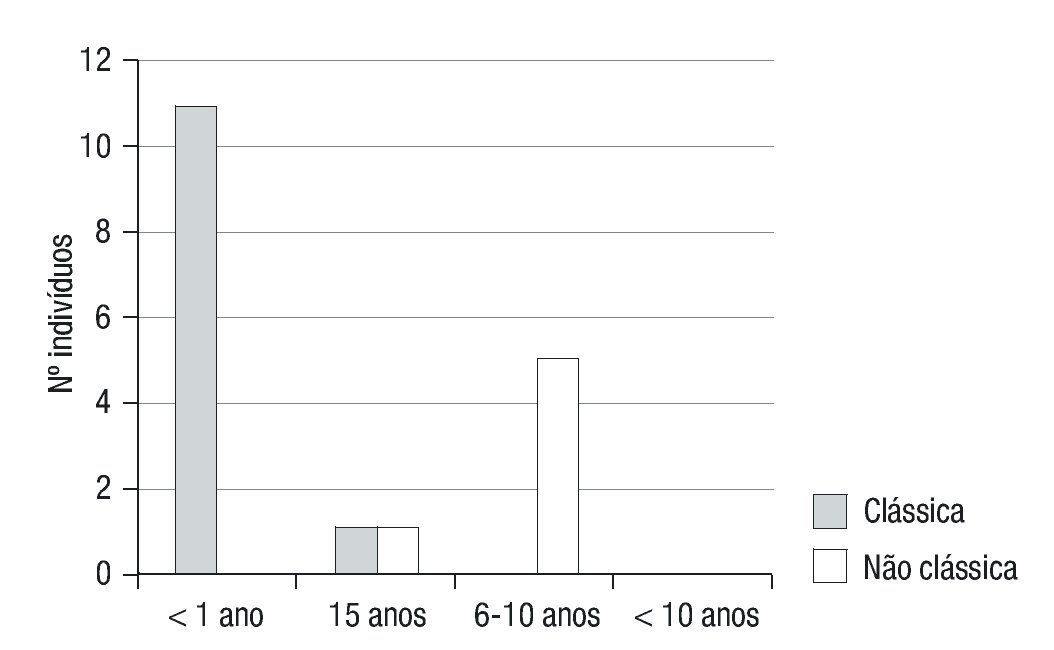
A apresentação clínica foi não-clássica em 6 doentes (33,3%) e clássica em 12 (66,7%) (forma CPS em 5 destes), com predomínio do sexo feminino (12/18) (Figura 1). A moda de idade no momento do diagnóstico foi inferior a 28 dias e de cerca de 6 anos nas formas clássicas (PS e VS) e não clássica, respetivamente (Figura 2).
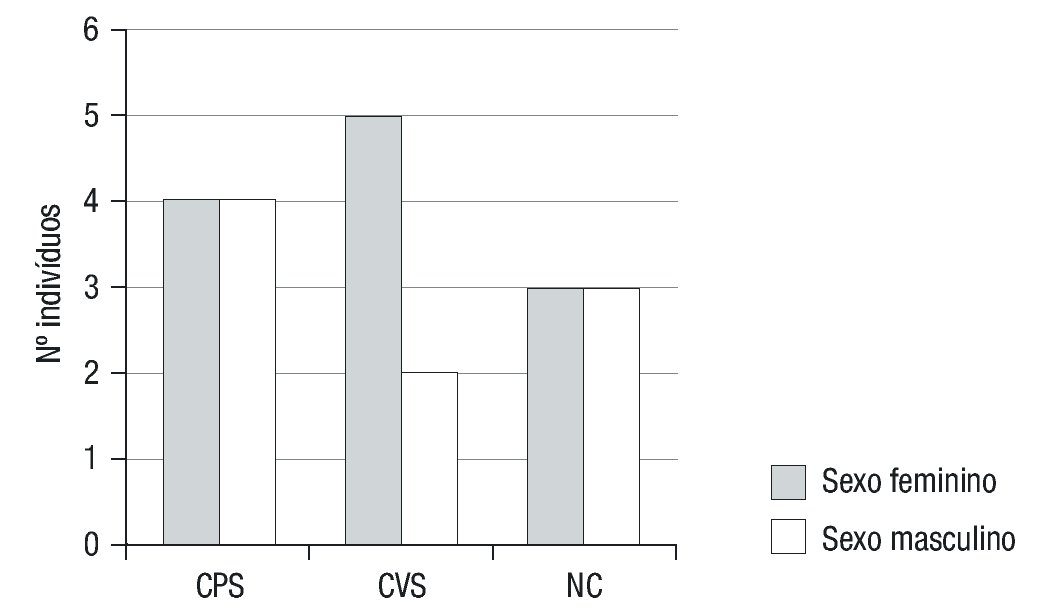
Figura 1. Distribuição por sexos de acordo com o fenótipo de apresentação da Hiperplasia Congénita da Suprarrenal. CPS: clássica perdedora de sal; CVS: clássica virilizante simples; NC: não clássica.
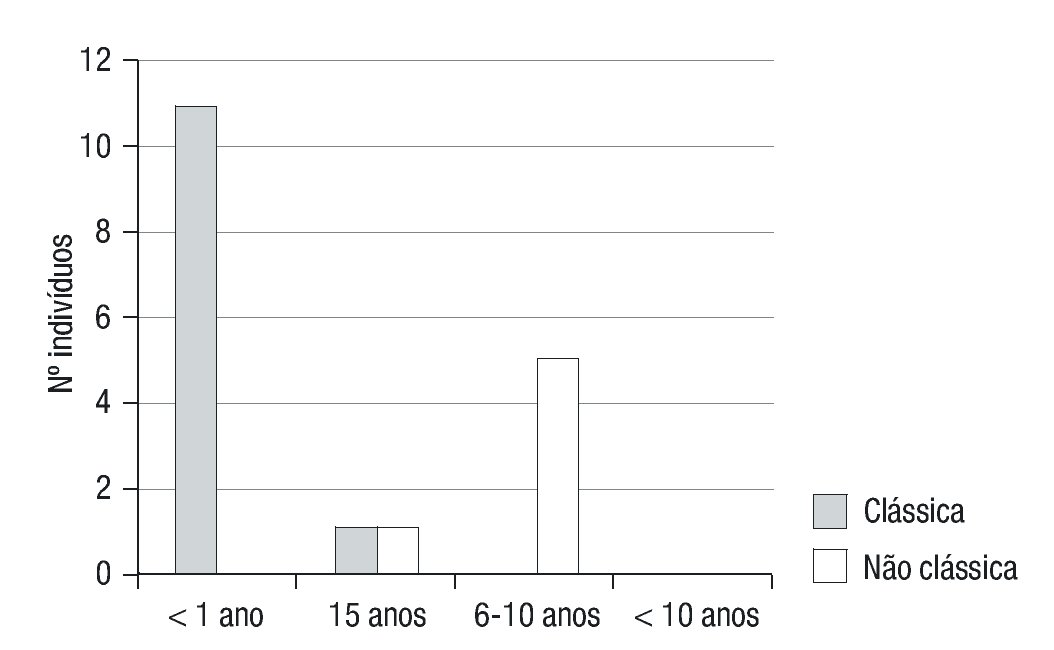
Figura 2. Faixa etária ao diagnóstico de Hiperplasia Congénita da Suprarrenal nas formas clássica (perdedora de sal e virilizante simples) e não-clássica.
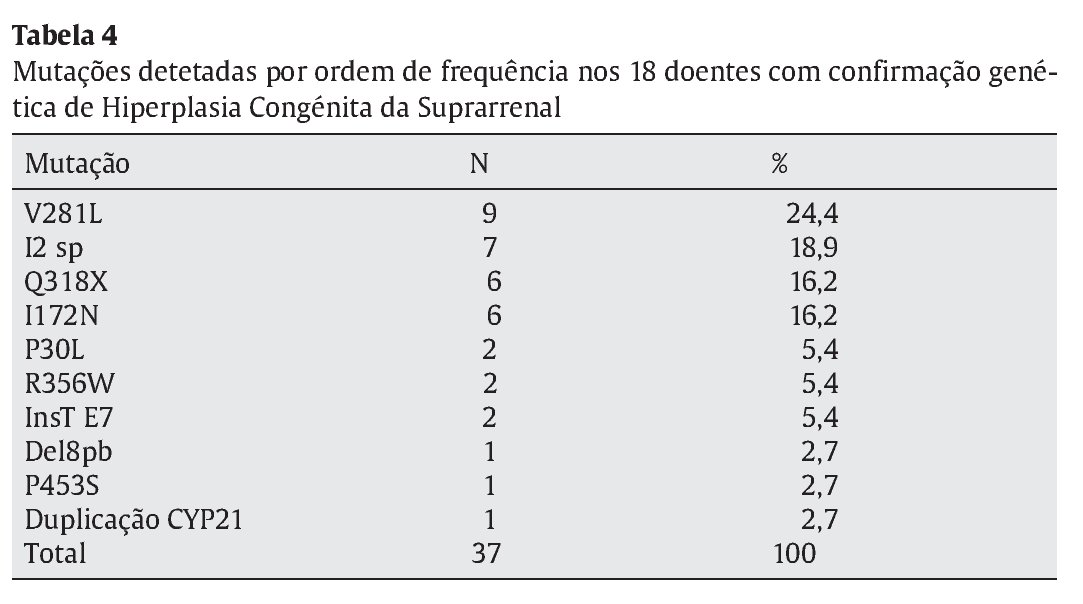
Identificaram-se 42 mutações, 37 destas em doentes com confirmação genotípica de HCSR. As 5 restantes corresponderam a mutações únicas, que portanto não suportam o diagnóstico molecular de HCSR. As mutações mais frequentemente identificadas foram a V281L, Q318X, I2 splicing e I172N (Tabela 4), responsáveis por 75,7% das mutações encontradas. As mutações foram homo--zigóticas em 39% dos casos, sendo as restantes heterozigóticas compostas.
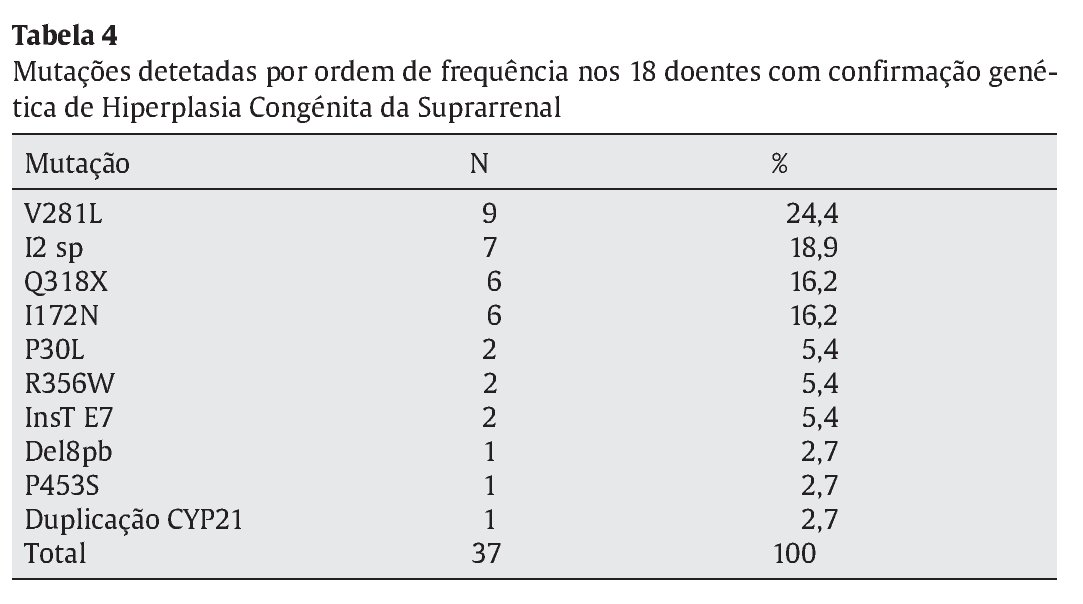
Dos 18 doentes incluídos no estudo realçam-se três pelas alterações genéticas detetadas e/ou pela apresentação clínica. O primeiro por apresentar três mutações no estudo molecular (V281L, Q318X, R356W) numa forma não clássica diagnosticada aos 3 anos de idade por um quadro de pubarca precoce. O segundo pelo facto de apresentar duplicação do gene CYP21A2, com uma mutação Q318X, numa menina com HCSR forma clássica (Prader III com uretra clitoriana) diagnosticada aos 5 anos de idade. Por último, o caso de um recém-nascido com um Prader V, internado ao 25o dia de vida por vómitos, má evolução ponderal, desequilíbrios hidro-eletrolítico e escroto vazio. O cariótipo foi 46XX e o estudo molecular confirmou 2 mutações no CYP21A2 (I2 splicing e InsT E7).
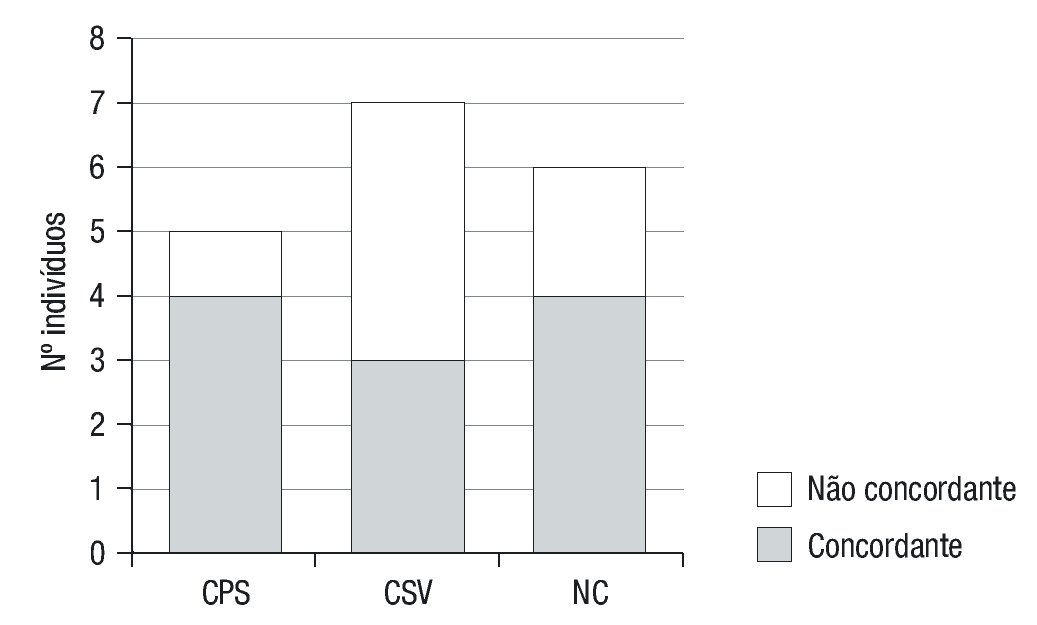
A apresentação fenotípica prevista foi concordante com a apresen--tação clínica da doença em 80% dos casos das formas clássicas PS, em 43% das formas clássicas VS e em 66,7% das formas não-clássicas (Tabela 5 e Figura 3). A concordância global foi de 61,1%.
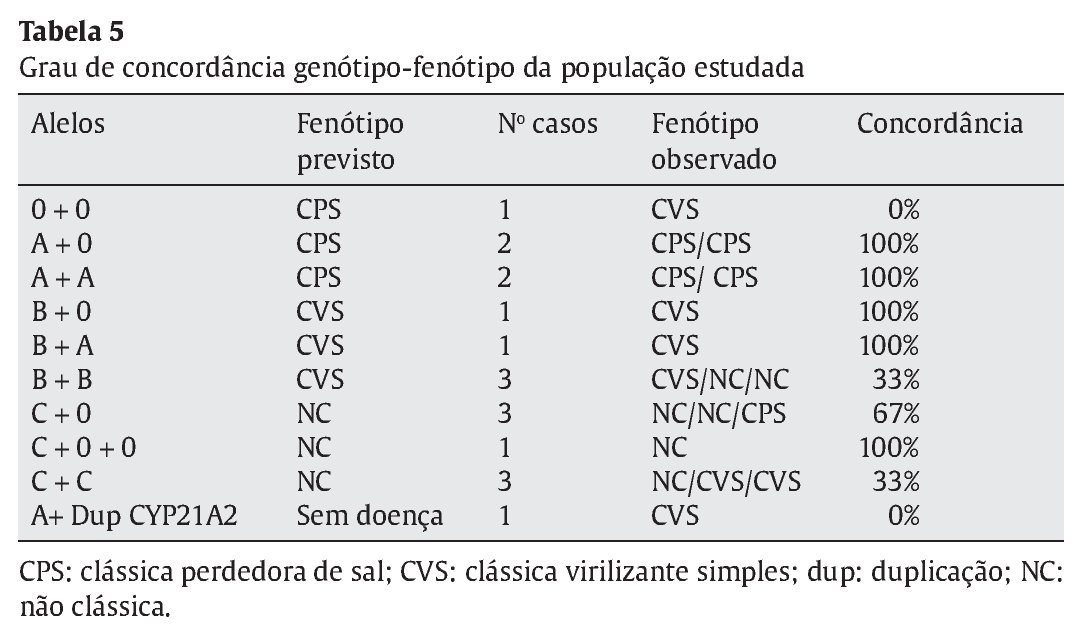
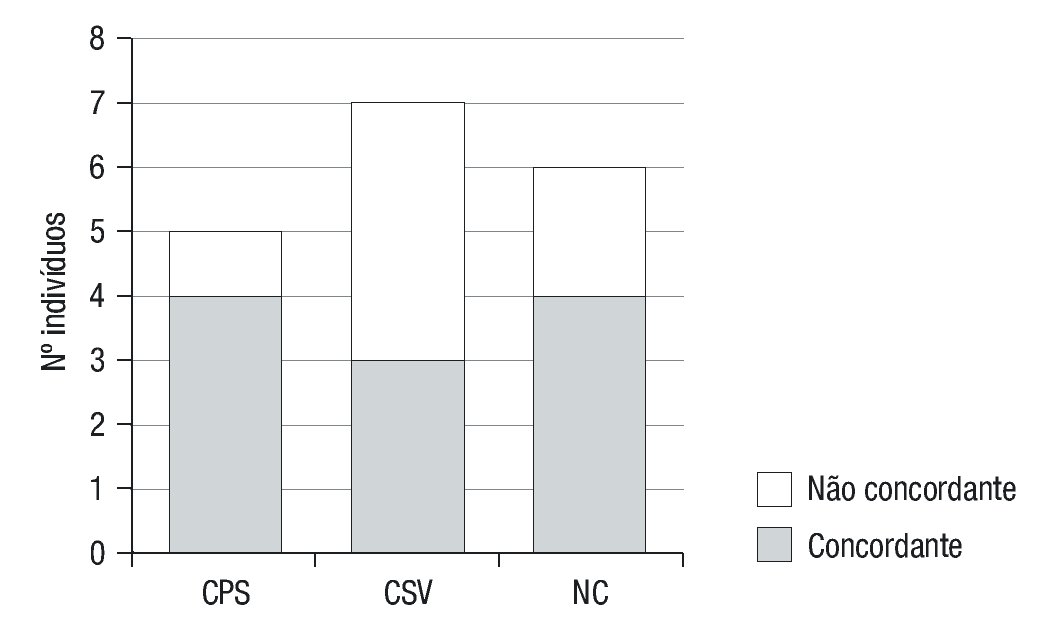
Figura 3. Concordância entre o fenótipo previsto e o observado. CPS: clássica perde-dora de sal; CVS: clássica virilizante simples; NC: não clássica.
Discussão
A classificação fenotípica da HCSR em forma clássica (PS ou VS) e não clássica é «teórica» podendo haver espectros que se sobrepõem na prática clínica.
Neste estudo evidencia-se uma proporção elevada de doentes do sexo feminino (66,7%), o que não seria esperado dado tratar-se de uma patologia autossómica recessiva, em que não é previsível haver assimetria na distribuição por sexos3. Esta divergência poderá estar relacionada com a pequena amostragem obtida, porém faz-nos pensar na possibilidade de maior dificuldade de diagnóstico no período neonatal na população masculina, dado que a virilização nestes pode ser subvalorizada. Do mesmo modo, a elevada proporção de formas clássicas (12/18) em relação às não clássicas, ao contrário do que seria de esperar, aler ta-nos para uma maior dificuldade no diagnóstico de formas com apresentação clínica mais ligeira7. Poderá existir uma menor valorização das manifestações clínicas, bem como dificuldade em estabelecer diagnóstico diferencial com a Síndrome do Ovário Poliquístico (SOP).
Em relação à pesquisa das mutações genéticas, verifica-se que o teste inicial que inclui as seis mutações mais frequentes identificou 86,5% (32/37) das mutações encontradas, parecendo-nos um bom método inicial de diagnóstico. Nos restantes cinco casos, em que o estudo inicial permitiu apenas a identificação de uma mutação, o diagnóstico molecular foi efetuado após sequenciação completa e análise por MLPA do gene CYP21A2. Nos doentes sem mutações identificadas não foi continuado o estudo, o que condicionará falsos negativos se existirem mutações apenas identificadas pela técnica MLPA. Não foram identificadas mutações novas.
Comparando com outros estudos, a nossa elevada percentagem de mutações homozigóticas e baixa diversidade de mutações, poderá ser explicada por algum grau de consanguinidade na amostra.
Dos casos destacados, o primeiro alerta para a importância de pesquisar sempre as mutações nos progenitores de forma a assegurar que a identificação de duas mutações corresponde à presença de ambos os alelos mutados e não apenas a um alelo com duas mutações. São pontualmente descritas em outros estudos estas mutações múltiplas6,9. O segundo caso demonstra que a associação de uma duplicação do CYP21 com um alelo mutado pode condicionar HCSR. Esta associação poderá de alguma forma interferir na atividade do gene, o que justificaria a apresentação clínica, apesar de não terem sido encontrados relatos semelhantes na literatura. Não pode contudo ser excluída a interferência de outros fatores moduladores da virilização10 ou a presença de mutações não identificadas11. O último caso relata uma virilização extrema no sexo feminino diagnosticada aos 25 dias de vida, com um fenótipo Prader V.
Apesar de 37 crianças terem um quadro clínico e analítico de HCSR, apenas em 18 foram encontradas mutações em ambos os alelos. No entanto, será sempre de considerar a possibilidade de existirem mutações não identificadas ou de ocorrer uma seleção genética diferente da teoricamente esperada, condicionando hiperandrogenismo e consequentemente manifestação de doença em indivíduos portadores de uma única mutação.
A concordância global genótio-fenótipo neste estudo foi de 61,1%: 80% nas formas clássicas PS, 43% nas clássicas VS e 66,7% das formas não-clássicas. Observou-se que os grupos com associações de mutações menos graves, nas formas clássica VS e NC (associação B+B e C+C respetivamente), apresentaram um valor mais elevado de discordância (77%). Dado o pequeno tamanho amostral, torna-se dificil a justificação destes resultados e a comparação com outros estudos, não deixando de ter interesse como uma base inicial para melhor conhecimento da nossa população.
Tal como descrito na literatura, a concordância genótipo-fenótipo foi notória nas formas de apresentação mais grave2,8, que condicionarão maior morbi-mortalidade neonatal. Dada a sua aplicabilidade na prática clínica5,6,9, nomeadamente no aconselhamento genético, os autores reforçam a importância da pesquisa das mutações do CYP21A2 para confirmação e caracterização da HCSR.
* Autor para correspondência.
Correio electrónico:natachafontes@gmail.com (N. Fontes).
INFORMAÇÃO DO ARTIGO
História do artigo:
Recebido a 13 de junho de 2012
Aceite a 2 de outubro de 2012
