




En este editorial nos gustaría presentar a los lectores de la Revista de Psiquiatría y Salud Mental algunos avances muy recientes que se han producido en el campo de la enfermedad de Alzheimer (EA) y el deterioro cognitivo leve (DCL), y que probablemente tendrán repercusiones de largo alcance en el diagnóstico y el tratamiento de este trastorno neurodegenerativo. Con objeto de orientar mejor al lector, conviene señalar que este editorial no es tanto una revisión científica (no entraremos en el detalle de los estudios individuales), como un presentación de una serie de hitos que el lector interesado puede utilizar como guía para profundizar más en este campo en rápida evolución. Intentaremos ser objetivos, aunque a veces tomaremos postura respecto a los avances que comentamos. Hay varias razones por las que un psiquiatra debe interesarse acerca de algunos de estos avances recientes en el campo de la EA y su pródromo. En primer lugar, por lo que respecta a la demografía, Norteamérica y Europa Occidental tienen poblaciones de edad cada vez más avanzada. Esto hace que la prevalencia de la EA esté aumentando y, por tanto, es probable que haya más pacientes que necesiten asistencia médica. En segundo lugar, la EA puede aportar enseñanzas que sean trasladables al campo de las enfermedades psiquiátricas, en el sentido de que tiene una arquitectura genética reproducible, biomarcadores con valor predictivo, un fenotipo celular y modelos animales para su estudio. No obstante, a pesar de estas ventajas, continúan existiendo problemas para la interpretación y el desarrollo de fármacos, y ello puede aportar información no sólo sobre lo que conviene hacer, sino también sobre lo que conviene no hacer.
Ensayos clínicosDurante el último año se han producido varios fracasos en ensayos clínicos realizados en la EA. En la evaluación de dimebon —un fármaco con un mecanismo de acción novedoso por cuanto estabiliza los poros mitocondriales—, los resultados fueron positivos en un ensayo realizado en Rusia. El fármaco fue desarrollado inicialmente como un antihistamínico y luego se observó que tenía propiedades de inhibición de la colinesterasa (y de ahí el interés inicial existente por el uso de este fármaco en la EA). Sin embargo, cuando se realizó un ensayo con fines de registro (Pfizer) en Europa Occidental y en Sudamérica y Norteamérica, los resultados fueron negativos1. No está claro por qué los dos ensayos produjeron resultados tan diferentes, pero sería razonable sospechar que el fármaco fuera menos activo neurobiológicamente de lo que al principio había parecido. Otra posible enseñanza extraída de este ensayo tiene menos que ver con la metodología clínica o con la farmacología que con una cuestión que a menudo se resalta al analizar la sociología de la ciencia, la llamada “maldición del ganador”2. Consiste en que el primer estudio positivo en un determinado campo puede deberse a factores o expectativas aleatorios y a menudo sus resultados no son reproducidos.
Más recientemente aún, un inhibidor de la gamma secretasa, semagacestat (Lilly), pareció causar un empeoramiento en los pacientes con EA que presentaban una demencia de leve a moderada, y ello motivó la interrupción prematura del ensayo (véase Alzforum.com). A pesar de que los cambios cognitivos en la fase 2 fueron bastante pequeños, se pasó a la fase 3 a partir de la evidencia obtenida en cuanto a biomarcadores. Este ensayo planteó ciertas dudas respecto a: a) el momento apropiado para la intervención (por ejemplo, si deben realizarse ensayos en momentos muy anteriores de la evolución de la enfermedad), y b) si la hipótesis del amiloide es correcta en cuanto al establecimiento de una diana para el tratamiento (recuérdese que se cree que la fragmentación de proteína precursora de beta amiloide (APP) por la acción del complejo de gamma secretasa produce especies moleculares de amiloide tóxicas, es decir, Abeta1-42 en determinadas circunstancias). Véase el artículo de Cummings3 para una visión interesante y más positiva del uso del amiloide beta como diana para el tratamiento.
BiomarcadoresJack et al4 han utilizado los resultados del estudio ADNI para proponer un concepto global de la forma en la que diferentes biomarcadores (por ejemplo, los volúmenes cerebrales regionales, los niveles de Abeta y tau en líquido cefalorraquídeo (LCR), el metabolismo cerebral de la glucosa, etc.) tienen diferentes tipos de valor predictivo en distintas fases de la enfermedad (por ejemplo, fase preclínica, DCL y neurodegeneración debida a la muerte celular en la enfermedad de Alzheimer en sí). Con independencia de sus detalles, algunos de los cuales son discutibles, el modelo ha resultado heurístico. El ADNI es un amplio consorcio multicéntrico (más de 50 centros) en el que grupos amplios de controles sanos de edad avanzada, individuos con DCL e individuos con EA leve son evaluados longitudinalmente con el empleo de pruebas cognitivas, exploraciones de neuroimagen estructurales, exploraciones de imagen con Pittsburgh B compuesto (PIB), tomografía por emisión de positrones (PET), fluorodesoxiglucosa (FDG) y exámenes de genoma completo. En todos los individuos incluidos se realizan exámenes sistemáticos y uniformes cada 6-12 meses. Los datos son de acceso público en www.loni.ucla; y se realizan con frecuencia volcados de dichos datos. En este modelo, un mal procesamiento de APP, que conduce a una agregación anormal de péptidos Abeta en forma de placas, constituye el evento inicial de la EA. Los biomarcadores de este proceso amiloide incluyen el aumento de la retención del trazador PIB (el PIB es un radioligando que se une al amiloide extracelular en el cerebro) y reducciones de Abeta en el LCR. La disfunción neuronal y la neurodegeneración pasan a ser entonces dominantes, como indica el aumento de los niveles de tau en LCR y las reducciones en la PET con FDG, sobre todo en las áreas parietotemporales. El deterioro cognitivo puede ser evidente, sobre todo en cuanto a la memoria episódica. Finalmente, se produce una atrofia cerebral regional, y en la EA en sí, esto está muy estrechamente relacionado con afectación de la función cognitiva. La cuestión clave en este concepto es que la importancia y la dinámica de los biomarcadores cambian a lo largo del tiempo (por ejemplo, el depósito de amiloide se estabiliza de forma temprana en el proceso de la enfermedad; la atrofia cerebral se acelera y es más informativa en las fases más avanzadas del proceso de la enfermedad). Sin embargo, nuestros propios análisis de los datos del ADNI y los estudios de nuestro laboratorio5 indican que la atrofia regional y los deterioros neurocognitivos, con la afectación consiguiente de la función cotidiana del paciente, se producen de forma relativamente temprana en el DCL.
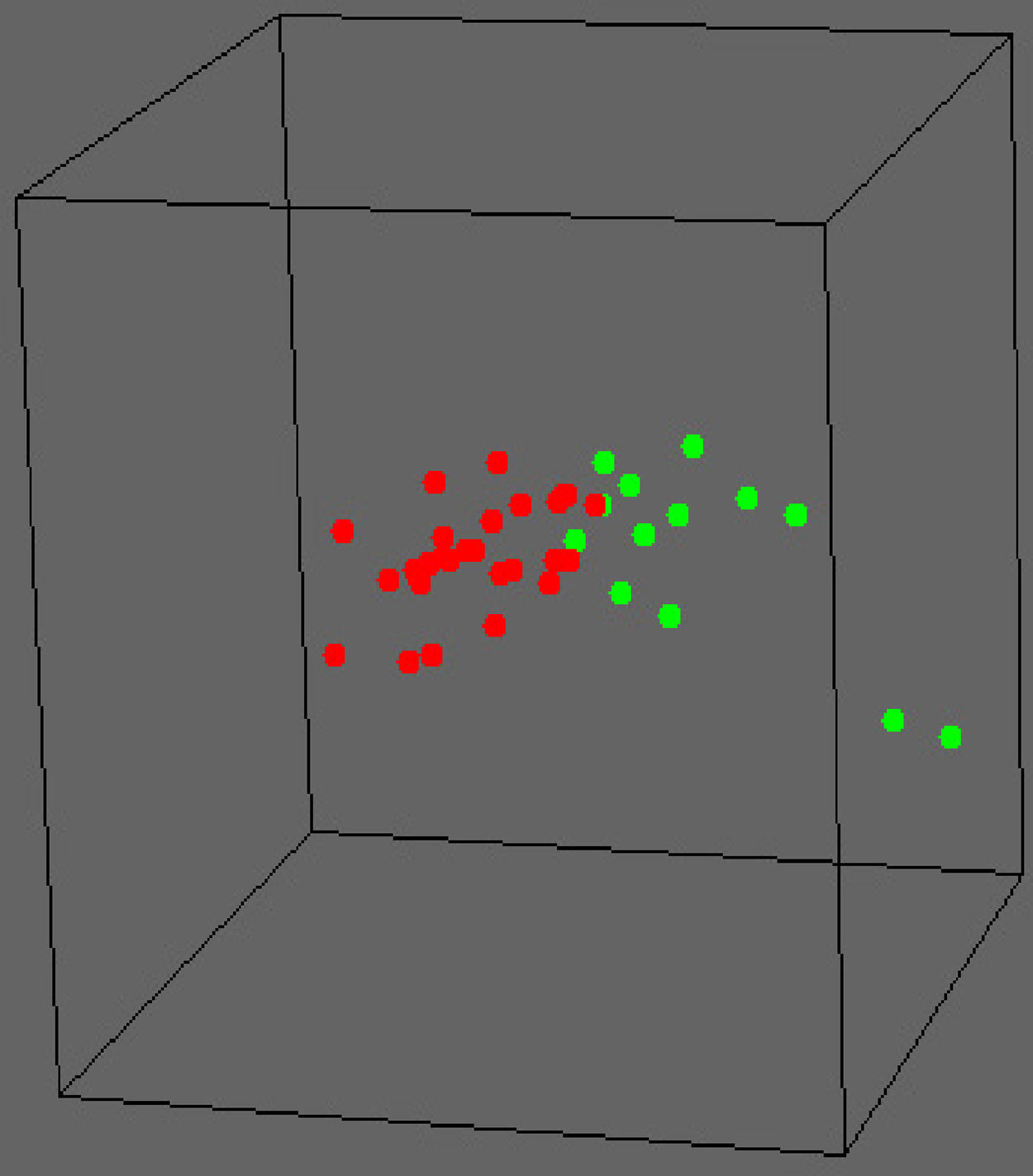
Cambios moleculares en el pródromo de la EALa EA es un trastorno neurodegenerativo caracterizado histopatológicamente por la presencia de placas neuríticas y ovillos neurofibrilares. Para identificar la neurosusceptibilidad y los factores neuroprotectores intrínsecos a nivel molecular, sin el efecto de confusión producido por las consecuencias patológicas posteriores, y orientarnos hacia la prevención, hemos estudiado la expresión en más de 26.000 genes con el empleo de la tecnología de microchips en muestras post mórtem de tejido cortical de 28 casos no portadores de APOE4 (el denominado grupo APOE3) y 13 casos portadores de APOE46. Dado que el genotipo APOE es el principal factor de riesgo genético para la EA de inicio tardío, con una odds ratio de 3,8 para la variante APOE4, el primero de estos grupos tenía un riesgo bajo de desarrollar la enfermedad, mientras que en el segundo grupo el riesgo de la enfermedad era alto. La media de edad en el momento de la muerte era de 42 años y ninguno de los cerebros presentaba una histopatología diagnóstica de EA al fallecer el paciente. Identificamos 70 transcriptos que diferían de forma significativa en los dos grupos, utilizando un nuevo método de “doble sustracción”. La diferenciación de los grupos fue significativa (fig. 1). Observamos también que múltiples vías biológicas Kyoto estaban alteradas en el grupo de APOE4, incluidas las involucradas en la función mitocondrial (los genes mostraban de manera uniforme una regulación negativa), la regulación del calcio y la reentrada en el ciclo celular. Con el empleo de análisis más sofisticados observamos entonces que estas moléculas formaban una red con múltiples conexiones entre sí y con APP y MAPT. Globalmente, nuestros resultados produjeron tres resultados principales:
- 1)
El estímulo más temprano para los procesos patogénicos en los individuos con E4 puede proceder de diversas anomalías en las cascadas de señalización y los procesos biológicos relativos a la regulación del calcio, la función mitocondrial, las anomalías de la regulación del ciclo celular, la apoptosis y la señalización Wnt. Varias de ellas han sido aspectos periféricos en los debates acerca de la patogenia de la EA; en nuestro estudio observamos que desempeñaban un papel central. Destacaba la ausencia en esta fase temprana del proceso de la enfermedad de la vía amiloide canónica, lo cual indica que esta última puede ser una cascada de procesos dentro de otra cascada.
- 2)
Puede haber procesos protectores activos, además de los procesos patológicos.
- 3)
Nuestros datos indican que hay un período prodrómico prolongado antes del inicio de la EA clínica, dado que la media de edad de muerte de los individuos de nuestra muestra era de 42 años.
.")
En resumen, la importancia de este estudio radica en que lo observado parecen ser los antecedentes del depósito de amiloide beta y la expresión de la fosfo-tau (es decir, lo observado en los ovillos neurofibrilares). Dado que el debate actual sobre la patogenia de la EA se centra en si las anomalías del amiloide beta preceden a la fibrilación de tau o viceversa, este estudio contribuye a señalar algunos mecanismos importantes que pueden preceder a ambos, y tener por tanto una relación causal en la cascada o cascadas moleculares largas que conducen a la EA clínica derivada de APOE4. Dado que este estudio se basó en el genotipo APOE, aporta una de las mejores perspectivas de que disponemos hasta el momento respecto a las alteraciones iniciales de la expresión génica que acompañan a la EA relacionada con el APOE4. No obstante, señalamos que no hay un consenso respecto a los mecanismos moleculares exactos a través de los cuales el APOE4 causa en última instancia la neurodegeneración.
Esta breve revisión debe leerse como un relato aleccionador. En un intento de mejorar los tratamientos psiquiátricos, sobre todo en la esquizofrenia, se han iniciado en este campo búsquedas muy amplias de resultados genéticos reproducibles, biomarcadores y modelos animales. Sin embargo, tal como ponen claramente de manifiesto los resultados de la bibliografía de la EA, incluso con estas ventajas traslacionales, la obtención de un tratamiento eficaz está lejos de ser automática.