





La literatura sobre los inductores en la epilepsia y el trastorno bipolar está contaminada por falsos negativos. Esta es la primera parte de una revisión exhaustiva de los fármacos antiepilépticas (FAE) con propiedades inductoras que utiliza los mecanismos farmacológicos y la medicina basada en la evidencia para aportar recomendaciones prácticas a neurólogos y psiquiatras sobre el modo de controlar sus efectos. La carbamazepina, el fenobarbital y la fenitoína son potentes inductores con efectos clínicos relevantes. Se calculan sus factores de corrección para los fármacos inducidos que han sido estudiados. Estos factores de corrección son meras simplificaciones para orientar a los clínicos, ya que entre las personas existe gran variedad en la intensidad de los efectos inductores. A medida que se publique nueva información, deberán modificarse los factores de corrección. Algunos de estos factores son tan elevados que algunos fármacos (el bupropión, la quetiapina o la lurasidona) no deberán administrarse conjuntamente con los inductores potentes. El clobazam, la eslicarbazepina, el felbamato, la lamotrigina, la oxcarbazepina, la rufinamida, el topiramato, la vigabatrina y el ácido valproico son inductores leves que pueden: a)ser inductores solo en dosis elevadas; b)combinar a menudo efectos inhibidores, y c)emplear meses en alcanzar sus efectos inductores máximos o su desaparición, e indudablemente necesitan más tiempo que los inductores potentes. Claramente los inductores potentes, y posiblemente los inductores débiles, tienen efectos relevantes en el metabolismo endógeno de: a)las hormonas sexuales; b)la vitaminaD; c)las hormonas tiroideas; d)el metabolismo lipídico, y e)el ácido fólico.
The literature on inducers in epilepsy and bipolar disorder is seriously contaminated by false negative findings. This is parti of a comprehensive review on antiepileptic drug (AED) inducers using both mechanistic pharmacological and evidence-based medicine to provide practical recommendations to neurologists and psychiatrists concerning how to control for them. Carbamazepine, phenobarbital and phenytoin, are clinically relevant AED inducers; correction factors were calculated for studied induced drugs. These correction factors are rough simplifications for orienting clinicians, since there is great variability in the population regarding inductive effects. As new information is published, the correction factors may need to be modified. Some of the correction factors are so high that the drugs (e.g., bupropion, quetiapine or lurasidone) should not co-prescribed with potent inducers. Clobazam, eslicarbazepine, felbamate, lamotrigine, oxcarbazepine, rufinamide, topiramate, vigabatrin and valproic acid are grouped as mild inducers which may (i)be inducers only in high doses; (ii)frequently combine with inhibitory properties; and (iii)take months to reach maximum effects or de-induction, definitively longer than the potent inducers. Potent inducers, definitively, and mild inducers, possibly, have relevant effects in the endogenous metabolism of (i)sexual hormones, (ii)vitaminD, (iii)thyroid hormones, (iv)lipid metabolism, and (v)folic acid.
En un editorial de 2 partes, en el que la partei1 se centra en la epilepsia y la parte ii2 en el trastorno bipolar, el autor propone la seria contaminación con falsos negativos de la literatura neuropsicofarmacológica acerca de las interacciones farmacológicas con los inductores metabólicos de los fármacos. Sistemáticamente se niegan, o al menos se subestiman, los efectos de los inductores, y la literatura publicada disponible resta importancia, de modo sistemático, a su relevancia clínica. Además, esta pauta de negación se produce tanto en la literatura sobre epilepsia1 como la publicada sobre el trastorno bipolar2, donde los inductores incrementan el metabolismo de muchos fármacos metabolizados por el citocromo P450 (CYP) y/o las enzimas uridina difosfato glucuronosiltransferasa (UGT). Este patrón de negación puede definirse como sistemático, ya que contamina las diferentes categorías farmacológicas: a)fármacos metabolizados por el CYP3A4; b)fármacos no metabolizados, y c)fármacos inductores leves1,2. Además, contamina la literatura de los tratamientos farmacológicos tanto de la epilepsia1 como del trastorno bipolar2, caracterizados por la polifarmacia, incluyendo el uso de inductores potentes tales como la carbamazepina o los inductores leves de introducción reciente, como la oxcarbazepina. Históricamente, la negación se produjo en primer lugar en la literatura acerca de los fármacos antiepilépticos (FAE)1, repitiéndose a continuación en la literatura sobre el trastorno bipolar2.
Este artículo de revisión trata de proporcionar una revisión exhaustiva de la literatura sobre los inductores de los FAE para para poner al día esta materia, en aras de aportar recomendaciones prácticas a los clínicos (partei). La parteii revisa los mecanismos farmacológicos que explican la inducción, y aporta información adicional para interpretar las interacciones farmacológicas de los FAE en el mundo real, lo que exige a menudo entender otros mecanismos farmacológicos además de dicha inducción.
La partei incluye secciones sobre: a)la búsqueda en la literatura (para las partesi yii); b)las limitaciones del uso de un enfoque práctico para proporcionar directrices a los clínicos sobre las interacciones farmacológicas de los inductores; c)los inductores potentes, incluyendo subsecciones sobre los 3 inductores más potentes: la carbamazepina, el fenobarbital y la fenitoína, una subsección breve sobre la primidona, y los efectos de estos potentes inductores sobre los principales tipos de fármacos; d)los inductores leves, con subsecciones sobre el clobazam, la eslicarbazepina, el felbamato, la lamotrigina, la oxcarbazepina, la rufinamida, el topiramato, la vigabatrina y el ácido valproico (AVP), y e)una descripción que los efectos inductores de los FAE van más allá de las interacciones farmacológicas e incluyen los efectos sobre el metabolismo de los compuestos endógenos.
Búsqueda en la literaturaEl autor ha venido realizando búsquedas en PubMed sobre esta cuestión durante más de 15años durante el proceso de publicación de: a)artículos de revisión en esta área3–9; b)estudios de los efectos de los inductores de los FAE en psiquiatría10–19, y c)un libro con directrices prácticas para cada FAE y/o agente estabilizador del estado de ánimo, con más de 1.000 referencias diferentes20. Todos ellos fueron publicados con ánimo de tratar de desarrollar un modelo sistemático de prescripción personalizada en psiquiatría21–26. En tal sentido, la incorporación o suspensión de un inductor es equivalente a la disminución o incremento de la dosis del sustrato, y se considera una forma de dosificación personalizada21. La recomendación en contra del uso de un inductor potente en el contexto del uso de un sustrato (p.ej., la fenitoína en un paciente que toma quetiapina) se considera una forma de selección personalizada del fármaco21.
Las amplias búsquedas de FAE individuales con propiedades inductivas realizadas en abril de 2011 fueron actualizadas para cada FAE en diciembre de 2013, para la publicación de este artículo. También se realizó una búsqueda en PubMed sobre la farmacocinética de la eslicarbazepina, ya que el autor no había completado las búsquedas previas sobre este compuesto antes de escribir este artículo.
Limitaciones de un enfoque práctico para la aportación de directrices a los clínicosEsta revisión va dirigida a los clínicos, en aras de resolver los problemas prácticos con los que se encuentran normalmente los psiquiatras y neurólogos, quienes tratan normalmente a pacientes sometidos a regímenes de polifarmacia. El autor trabaja como asesor del sistema público de salud mental de Kentucky, y su experiencia diaria es que los psiquiatras y neurólogos no son conscientes de que los inductores de los FAE ejercen una gran influencia sobre la dosificación de otros fármacos, y y tienen una comprensión limitada sobre el modo de corregir estos efectos. Esta es una revisión para los clínicos en ejercicio, y puede no satisfacer completamente a los lectores con una orientación científica. En el campo de la psicofarmacología trabajan 2 grandes grupos de científicos: los científicos básicos, que se centran en los mecanismos farmacológicos, y los que adoptan un enfoque estadístico, el enfoque denominado medicina basada en la evidencia (MBE)27.
Enfoque de los mecanismos farmacológicosLos farmacólogos que trabajan en un laboratorio pueden debatir28 sobre la clasificación práctica de los inductores potentes y leves utilizada en este artículo de revisión. Smith et al.28 puntualizarían que todos los inductores de los FAE, independientemente de su potencia, utilizan los mismos mecanismo de inducción y desde el punto de vista farmacológico, la potencia puede definirse como la afinidad del inductor hacia la enzima inducida. Esto es evidentemente correcto desde un punto de vista farmacológico, pero nuestra capacidad de extrapolar al entorno clínico nuestros conocimientos de los mecanismos farmacológicos que explican la inducción es bastante limitada. Nuestros conocimientos son insuficientes para elaborar una clasificación simplificada destinada a los clínicos, cuyo interés fundamental es el tratamiento de sus pacientes. En dicho sentido, la revisión de nuestros limitados conocimientos de los mecanismos farmacológico que explican la inducción se describe en la parteii de este artículo, que puede ser consultada por los clínicos con un interés adicional. Como no se han estudiado sistemáticamente muchas interacciones farmacológicas específicas, y no se dispone de información que oriente a las decisiones clínicas, el autor debe extrapolar a menudo, en su propia práctica clínica, otros fármacos con un perfil metabólico similar, siguiendo los principios de los mecanismos farmacocinéticos. Sin embargo, el autor ha aprendido a ser extremadamente cauto a la hora de extrapolar otras interacciones farmacológicas, ya que la literatura tiene una predisposición sistemática en contra de los efectos inductivos de los FAE, y está frecuentemente contaminada con afirmaciones incorrectas que se van repitiendo de un artículo a otro. Los mejores ejemplos de las afirmaciones incorrectas sobre los efectos de los inductores se refieren al topiramato entre los FAE, y a la paliperidona entre los fármacos utilizados para el trastorno bipolar.
Muchos artículos nos recuerdan que el topiramato se elimina inalterado en la orina, aunque en la realidad es parcialmente metabolizado (la cifra normalmente citada es del 20%) por los CYP29. Hasta donde alcanza el conocimiento del autor, nunca se han estudiado los CYP específicos. Desafortunadamente, la literatura no recalca que el metabolismo en situaciones normales no es el mismo que el metabolismo bajo inducción. Aunque todavía no se ha estudiado bien el metabolismo del topiramato bajo inducción, es claro que la eliminación del topiramato se incrementa al doble con la administración de carbamazepina o fenitoína, lo que demanda la duplicación de la dosis29. Por tanto, cuando se prescriben inductores potentes, se metaboliza una proporción mayor de topiramato, probablemente de alrededor del 40%. El prospecto de paliperidona describe una disminución del 37% del área de paliperidona bajo la curva (AUC) tras la ingesta de 400mg/día de carbamazepina durante un periodo no especificado. Dicho periodo ha sido descrito recientemente como de 3semanas30, lo que es insuficiente para alcanzar la inducción máxima de la carbamazepina. Durante años, basándose en su experiencia con la risperidona10,11,13, el autor31 había sostenido la hipótesis de que el metabolito principal de la risperidona, comercializado como paliperidona, que la compañía farmacéutica describía que no se metabolizaba podría ser parecido al topiramato y bastante susceptible a la inducción. Recientemente, Yasui-Furukori et al.32 han demostrado que la hipótesis del autor era correcta; la administración de 600mg/día de carbamazepina durante 2-4semanas se asociaba con una reducción media de las concentraciones plasmáticas de paliperidona a su tercera parte. Esto requeriría la triplicación de la dosis de paliperidona en estos pacientes. Es posible que se precise un factor de corrección de la paliperidona aún mayor en pacientes con prescripción de dosis superiores de carbamazepina, o con duraciones de tratamiento mayores.
Enfoque de la medicina basada en la evidenciaLos científicos con un enfoque de la MBE estricto no estarán nunca satisfechos con las recomendaciones aportadas en este artículo de revisión, ya que muchas de ellos se basan a menudo en los reportes de casos, o en extrapolaciones basadas en lo que se conoce como mecanismos farmacocinéticos de los fármacos con un metabolismo similar. Ignorar el valor de la extrapolación puede tener consecuencias perjudiciales. Basándose en la extrapolación que la oxcarbazepina puede ser un inductor menos potente que la carbamazepina, el autor propuso que la oxcarbazepina tiene efectos inductivos sobre la lamotrigina. Esto ayudó a detectar en la vida real 2 casos de síndrome de Stevens-Johnson12 inicial secundarios a la suspensión de oxcarbazpine, evitando la progresión de una reacción adversa medicamentosa (RAM) potencialmente letal.
Los enfoques de la MBE están naturalmente limitados en la literatura sobre inductores ya que, como ha descrito el autor1,2, la literatura actual está contaminada por una predisposición sistemática a la negación de la relevancia clínica de los inductores de los FAE. Es muy fácil diseñar un ensayo clínico aleatorizado (ECA), utilizando inductores de los FAE, que produzca unos resultados negativos; solo hay que prescribir los inductores utilizando dosis o tiempos por debajo de los necesarios para producir la inducción. El resultado será un ensayo negativo, pudiéndose concluir que el inductorA no tiene un efecto sobre el sustratoB1,2. Lamentablemente, las empresas farmacéuticas han diseñado pocos ECA para el estudio de las interacciones farmacológicas en situaciones clínicamente relevantes, en aras de aportar recomendaciones a los clínicos.
Este autor podría entablar entonces un debate sobre los errores tipo en estadística, que sería relevante para el enfoque de la MBE, aunque prefiere reconocer que no vivimos en un mundo ideal. En dicho mundo ideal sería preferible seguir los principios de la MBE, realizando cientos de ECA. Dichos ensayos aportarían recomendaciones para los clínicos acerca de la adecuada prescripción conjunta de los inductores de los FAE en situaciones de polifarmacia. Pero la realización de cientos de ECA sería bastante costosa y requeriría un esfuerzo masivo. En el mundo real, que incluye una ausencia de financiación suficiente en esta área, el autor ha utilizado muestras ya recolectadas, para explorar los efectos inductivos. El resultado más satisfactorio ha ocurrido en los estudios del autor sobre los efectos del AVP en el metabolismo de la clozapina. En el primer estudio14, un modelo matemático sofisticado, demostró que el AVP puede ser un inductor del metabolismo de la clozapina utilizando una muestra de conveniencia compuesta de un total de 415 muestras plasmáticas de clozapina recolectadas para otros fines. Dichas muestras se obtuvieron para realizar una monitorización terapéutica (MT) en 83 pacientes remitidos por sus terapeutas para seguimiento clínico y en 172 pacientes de estudios previos de las interacciones farmacológicas14. Lamentablemente, el mismo tipo de modelo matemático no fue capaz de demostrar los efectos inductivos del AVP en la olanzapina en una muestra de conveniencia que incluía un total de 360 muestras plasmáticas de olanzapina15. Las muestras se obtuvieron combinando la MT de 116 pacientes y los estudios de interacción farmacológica de 47 pacientes15. La falta de demostración de la inducción del AVP en el metabolismo de la olanzapina fue bastante sorprendente, puesto que el metabolismo de la olanzapina y la clozapina son bastante similares. Para detectar los efectos inductivos del AVP se precisó un diseño más sofisticado en un estudio prospectivo de la olanzapina mucho más pequeño (18 pacientes, 3 muestras por paciente, un total de 46 muestras de olanzapina), que, demostró los efectos inductivos del AVP sobre el metabolismo de la olanzapina16. Desafortunadamente, dicho estudio prospectivo planteó más cuestiones, al indicar que el AVP puede también inhibir de modo competitivo el metabolismo de la olanzapina, y que la duración del AVP puede ser importante a la hora de determinar los efectos netos (inducción frente a inhibición)17. De hecho, es posible que las 4 semanas de tratamiento con AVP en este estudio prospectivo no fueran suficientes para alcanzar la inducción máxima del metabolismo de la olanzapina.
Aunque el párrafo anterior resalta la necesidad de mayores niveles de evidencia en cuanto a las interacciones farmacológicas, el desarrollo total de un enfoque de la MBE puede no resolver estas cuestiones en todos los pacientes. Los estudios bien controlados tienden a centrarse en los pacientes promedio, pero en el mundo clínico muchos pacientes son lo que los estadísticos llaman valores atípicos o extremos que no siguen los patrones promedios ni están bien representados por la media. El autor no sabe si hay pacientes que son completamente resistentes a los inductores y que no demuestren una inducción clínicamente relevante en presencia de inductores potentes con dosis y tiempos probables para causar dicha inducción, aunque no se sorprendería de que existieran. Pero tiene amplia experiencia con los pacientes relativamente raros (probablemente <1%) que aparentan tener respuestas muy potentes a los inductores. En la tabla 1 se describen brevemente algunos de ellos, ya que la literatura raramente describe su existencia1.
Listado de pacientes con sensibilidad extrema a los efectos inductivos
| Caso 1. Paciente ♂ caucásico hospitalizado con esquizofrenia y epilepsia de inicio tardíoa |
| A) Sensibilidad extrema a la inducción de CYP3A4 |
| - Carbamazepina: anteriormente fue necesario administrar 1.500-2.000mg/díab para alcanzar las concentraciones séricas necesarias |
| - QuetiapinaCon fenitoína y AVP, el ratio C/D de quetiapina fue 10 veces menor a lo esperado |
| Con AVP, el ratio C/D de quetiapina fue 2-6 veces inferior a lo esperado |
| - DiazepamcCon fenitoína, las concentraciones séricas fueron indetectables a pesar de tomar 30mg/día |
| Con AVP, la eliminación de diazepam tras 30mgi.m. fue ≥3 superior a lo esperado |
| B) Posible autoinducción del AVP al tomar concentrados, aunque no presente en divalproex sódico |
| Con concentrado, se necesitaron 5.250mg/díad para alcanzar las concentraciones terapéuticas |
| Con divalproex sódico fueron suficientes 2.000mg/díae para lograr concentraciones terapéuticas |
| C) El metabolismo de clozapina y olanzapina (fármacos CYP1A2) fue normal para un varón fumadorf |
| Caso 2. Paciente ♂ afroamericano hospitalizado con trastorno esquizoafectivog |
| A) Inducción extrema de la clozapina por el AVP (divalproex sódico) |
| Fumador (20 cigarrillos/día): se precisó administrar ≥650mg/día para alcanzar concentraciones séricas >350ng/mlh |
| Fumador y AVP: se precisó administrar ∼1.200mg/día para alcanzar concentraciones séricas >350ng/mli |
| Durante la inducción del tabaco, el paciente demostró una elevada capacidad metabólica para la clozapina |
| La adición de la inducción del AVP originó la mayor capacidad metabólica de la clozapina que el autor haya visto nunca |
| Caso 3. Paciente ♂ caucásico hospitalizado con trastorno esquizoafectivoj |
| A) Sensibilidad extrema a la inducción de CYP3A4 |
| -Carbamazepina: se necesitaron hasta 2.800mg/díab para alcanzar concentraciones séricas terapéuticas; esto se explica parcialmente por los depósitos de tejido graso y el alto volumen de distribuciónk |
| - RisperidonaCon carbamazepina, aproximadamente 4 veces superior a la capacidad metabólica normal |
| Con divalproex sódico el metabolismo de la risperidona era normal |
| - Paliperidona: con carbamazepina, alta capacidad de metabolizar la paliperidona |
| B) Inducción extrema de olanzapina por AVP (divalproex sódico) y omeprazol |
| -Olanzapina: con divalproex sódico y omeprazol, 1,5-2 veces superior al metabolismo normal |
| Caso 4. Paciente ♂ caucásico hospitalizado con trastorno bipolarl |
| A) Posible autoinducción de VPA al administrarse divalproex sódico |
| – Dado de alta con 4.000mg/díam |
| Caso 5. Paciente ♂ hospitalizado con epilepsia en el lóbulo frontal y temporal y psicosisn |
| A) Sensibilidad extrema a la inducción de UGT durante el tratamiento con fenitoína y fenobarbital |
| -Lamotrigina: se precisó una dosis 2,6 veces superior a la recomendadao |
| -Lorazepam: se toleraron dosis >20mg/día sin sedación |
| Caso 6. Paciente ♂ hospitalizado con esclerosis tuberosa y tumores renalesp |
| A) Incremento de la capacidad metabólica de AVP al administrarse fenitoína y VPA |
| -Divalproex sódico: incremento de la dosis >10.000mg/día para alcanzar las concentraciones séricas terapéuticasq |
| -Fenitoína: sin cambios en el metabolismo de la fenitoína y sin necesidad de modificar la dosificación |
Dosis habituales de carbamazepina para alcanzar las concentraciones terapéuticas: 800-1.200mg/día. Dosis máxima recomendada: 1.600mg/día124.
El diazepam se metaboliza principalmente por CYP2C19; CYP3A4 es una enzima auxiliar. En este paciente, CYP3A4 era probablemente la enzima metabólica principal para el diazepam.
Los datos fueron publicados sin explicación farmacológica125. El ratio C/D de AVP fue de 0,013-0,017.
Los datos fueron publicados sin explicación farmacológica125. El ratio C/D de AVP fue de 0,036-0,048.
En el momento de la dosis máxima de carbamazepina, el IMC era de 40, con un peso corporal de 191kg. La elevada dosis se debe parcialmente a la obesidad128.
El ratio C/D de AVP fue de 0,024-0,033 durante el primer mes, disminuyendo a 0,017-0,018 durante el segundo mes.
El paciente necesitó 1.600mg/día para alcanzar las concentraciones séricas terapéuticas de lamotrigina. La dosis máxima recomendada es de 600mg/día.
Seguimiento durante 4años (edad de 44 a 48años hasta que falleció). Inicialmente tenía angiomiolipomas en ambos riñones. Durante el segundo año, una masa creciente en el riñón derecho condujo al diagnóstico de un posible carcinoma renal, y el resultado patológico de la nefrectomía sugirió un angiomiolipoma. Al tercer año, la metástasis cerebral se hizo evidente.
Al principio, cuando el paciente tenía tumores renales bilaterales, precisó cerca de 5.000mg/día de divalproex sódico para alcanzar las concentraciones terapéuticas, con ratios C/D de AVP de 0,010-0,018. Tras la nefrectomía, una vez que la presencia del cáncer renal metastásico era obvia, precisó 10.500mg/día de divalproex para alcanzar las concentraciones terapéuticas, con ratios C/D de AVP de 0,005-0,009.
Al revisar la literatura, el autor ha tratado de combinar los enfoques de los mecanismos farmacológicos y la MBE, en un intento de obtener las mejores recomendaciones para los clínicos en su práctica clínica. La reinterpretación del potencial de las interacciones farmacológicas de la vigabatrina y el clobazam pueden ser el mejor ejemplo de este pensamiento combinatorio del autor. Ambos fármacos fueron utilizados en Europa durante muchos años, y no fueron considerados inductores. Su introducción en Estados Unidos requirió que la Food and Drug Administration (FDA) obligara a realizar estudios in vitro, en relación con su potencial de interacción farmacológica, que demostraron que ambos fármacos son inductores leves. Esto llevó al autor1 a revisar los antiguos ECA que, efectivamente, sugerían que dichos fármacos eran inductores leves, aunque los artículos antiguos ignoraban —o incluso rechazaban— la posibilidad de que ambos fármacos fueran inductores. La vigabatrina y el clobazam nos enseñan que la literatura en esta área es bastante poco fiable. Basándonos en las limitaciones de la literatura antigua, podemos pronosticar con facilidad que en 5años este artículo de revisión puede estar obsoleto y precisar modificaciones amplias, incluyendo las modificaciones de los factores de corrección para la dosificación.
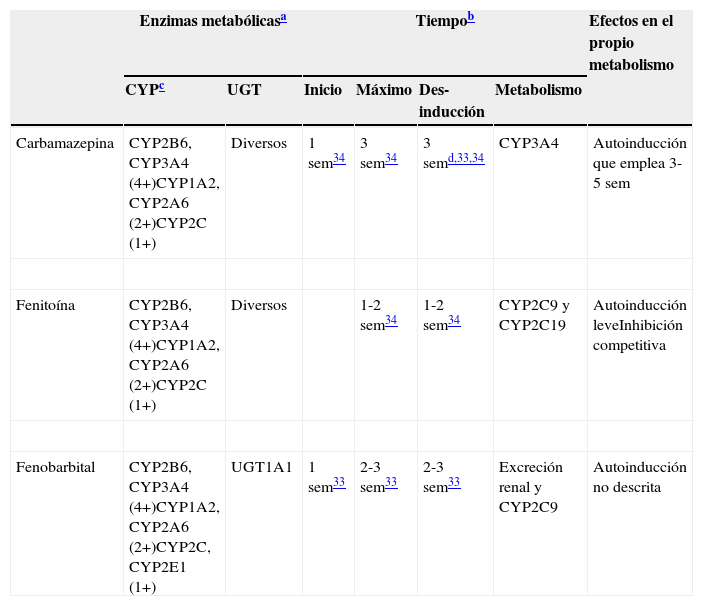
Inductores potentesExiste un consenso general en la literatura33,34 acerca de que tres FAE, carbamazepina, fenobarbital y fenitoína, son inductores clínicamente relevantes. En realidad, se piensa que todos los barbitúricos son inductores, aunque esta revisión menciona solo brevemente a otro de ellos, la primidona. La tabla 233–36 proporciona a los clínicos un resumen de las características comunes de estos potentes inductores. Los clínicos que estén interesados en una mejor comprensión de los diversos CYP y UGT, así como de la relevancia de su inducción, están invitados a consultar la tabla 3. Alguna información indica que es posible que la fenitoína pueda ser un inductor más promiscuo y potente que la carbamazepina, ya que se ha reportado que la adición de fenitoína puede precisar la duplicación de la dosis de carbamazepina37, lo que indica que la fenitoína impulsa la inducción, más allá de la autoinducción propia de la carbamazepina. Como el fenobarbital se utiliza actualmente con menos frecuencia en los países desarrollados, existe información limitada acerca de sus propiedades inductivas en muestras clínicas.
Características de los inductores potentes
| Enzimas metabólicasa | Tiempob | Efectos en el propio metabolismo | |||||
|---|---|---|---|---|---|---|---|
| CYPc | UGT | Inicio | Máximo | Des-inducción | Metabolismo | ||
| Carbamazepina | CYP2B6, CYP3A4 (4+)CYP1A2, CYP2A6 (2+)CYP2C (1+) | Diversos | 1 sem34 | 3 sem34 | 3 semd,33,34 | CYP3A4 | Autoinducción que emplea 3-5 sem |
| Fenitoína | CYP2B6, CYP3A4 (4+)CYP1A2, CYP2A6 (2+)CYP2C (1+) | Diversos | 1-2 sem34 | 1-2 sem34 | CYP2C9 y CYP2C19 | Autoinducción leveInhibición competitiva | |
| Fenobarbital | CYP2B6, CYP3A4 (4+)CYP1A2, CYP2A6 (2+)CYP2C, CYP2E1 (1+) | UGT1A1 | 1 sem33 | 2-3 sem33 | 2-3 sem33 | Excreción renal y CYP2C9 | Autoinducción no descrita |
4+: inducción masiva; 2+: inducción moderada; 1+: inducción leve; sem: semanas.
Constituyen tiempos aproximados incluidos en los artículos de revisión29,30. Los lectores deben ser conscientes de que se han realizado pocos estudios para verificar estos tiempos.
No todos los CYP tienen la misma capacidad de ser inducidos por los inductores potentes. Se incluyen más detalles en la tabla 3. Se piensa que los inductores potentes tienen efectos masivos (4+) sobre CYP2B6 y CYP3A4. Por otro lado, los inductores potentes tienen efectos únicamente leves sobre la subfamilia CYP2C, que incluye a CYP2C8, CYP2C9 y CYP2C1935. Aunque la literatura no es específica en este punto, el autor piensa que CYP1A2 puede ser inducido de modo intermedio entre los efectos potentes sobre CYP2B6 y CYP3A4 y los efectos leves sobre la subfamilia CYP2C, describiéndose como moderado (2+). Existe información limitada sobre CYP2A6, que sugiere un potencial de inducción moderada (2+), aunque los clínicos deben ser conscientes de que pocos fármacos son metabolizados por CYP2A6; esta es la ruta metabólica principal de la nicotina. Existe poca información acerca de CYP2E1, que puede tener un potencial leve para la inducción. Los clínicos deben ser conscientes de que pocos fármacos son metabolizados por CYP2E1, aunque es una ruta metabólica menor del alcohol y ciertos fármacos antiepilépticos.
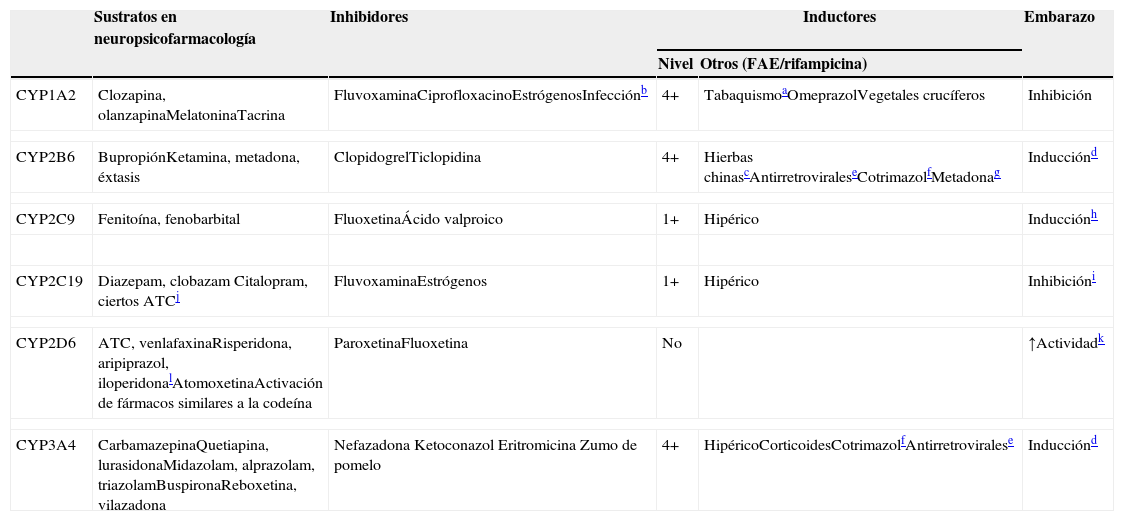
CYP implicadas en el metabolismo de los fármacos
| Sustratos en neuropsicofarmacología | Inhibidores | Inductores | Embarazo | ||
|---|---|---|---|---|---|
| Nivel | Otros (FAE/rifampicina) | ||||
| CYP1A2 | Clozapina, olanzapinaMelatoninaTacrina | FluvoxaminaCiprofloxacinoEstrógenosInfecciónb | 4+ | TabaquismoaOmeprazolVegetales crucíferos | Inhibición |
| CYP2B6 | BupropiónKetamina, metadona, éxtasis | ClopidogrelTiclopidina | 4+ | Hierbas chinascAntirretroviraleseCotrimazolfMetadonag | Inducciónd |
| CYP2C9 | Fenitoína, fenobarbital | FluoxetinaÁcido valproico | 1+ | Hipérico | Inducciónh |
| CYP2C19 | Diazepam, clobazam Citalopram, ciertos ATCj | FluvoxaminaEstrógenos | 1+ | Hipérico | Inhibicióni |
| CYP2D6 | ATC, venlafaxinaRisperidona, aripiprazol, iloperidonalAtomoxetinaActivación de fármacos similares a la codeína | ParoxetinaFluoxetina | No | ↑Actividadk | |
| CYP3A4 | CarbamazepinaQuetiapina, lurasidonaMidazolam, alprazolam, triazolamBuspironaReboxetina, vilazadona | Nefazadona Ketoconazol Eritromicina Zumo de pomelo | 4+ | HipéricoCorticoidesCotrimazolfAntirretroviralese | Inducciónd |
4+: inducción masiva; 2+: inducción moderada; 1+: inducción leve; ADT: antidepresivo tricíclico; FAE: fármaco antiepiléptico.
Los hidrocarburos aromáticos policíclicos del humo tienen efectos inductivos. Estos compuestos se encuentran también en los alimentos a la parrilla y los granos del café tostado, que pueden tener también efectos inductivos129.
Las infecciones respiratorias, otras infecciones graves, tales como la pielonefritis o apendicitis, o incluso las inflamaciones grandes, pueden inhibir CYP1A2, porque las citoquinas liberadas inhiben CYP1A2.
El ferulato de sodio se comportó como un inductor del metabolismo del bupropión en un estudio. Se trata de la sal sódica del ácido ferúlico, que está ampliamente distribuida en hierbas y fórmulas chinas como Ligusticum, Chuanxiong y Chaihu-Sugan-San130. Otro inductor es la baicalina, un glucurónico flavonoide extraído de la planta médica Radix scutellariae, que está presente en frutas, vegetales y bebidas derivadas de las plantas (té, vino tinto), y gran variedad de hierbas medicinales, que incluyen: Huang-Lian-Jie-Du-Tang, hangeshashinto, San-Huang-Xie-Xin-Tang, Da-Chai-Hu-Tang y Xiao-Chai-Hu-Tang131.
El embarazo induce definitivamente a CYP2B6 y CYP3A4 132. Conforme a un estudio in vitro, CYP2B6 y CYP3A4 son inducidas tanto por los estrógenos como por la progesterona. La progesterona induce también a CYP3A5.
Los estudios in vitro indican que la metadona puede inducir su propio metabolismo. Se cree que este efecto está mediado no solo por CYP2B6 sino también por CYP3A4 138.
Se piensa que CYP2C9 se incrementa durante el embarazo debido al incremento de la eliminación de fenitoína. No puede descartarse que los mecanismos diferentes a la inducción de CYP2C9 puedan explicar los cambios en la eliminación de la fenitoína durante el embarazo. El estradiol incrementa la actividad de CYP2C9 sin afectar a la expresión, mediante mecanismos desconocidos139.
Se piensa que los estrógenos son inhibidores competitivos de CYP2C19, aunque un estudio reciente sugería que pueden inhibir la expresión de CYP2C19 140.
CYP2C19 es la enzima principal para la demetilación de amitriptilina, clomipramina e imipramina. A continuación sus metabolitos son metabolizados mediante hidroxilación, principalmente por CYP2D6.
No se comprende bien la causa del posible incremento de la actividad de CYP2D6 durante el embarazo, ya que se piensa que CYP2D6 no puede ser inducida. Un estudio reciente sugería que el embarazo puede eliminar un supresor de la expresión de CYP2D6 141.
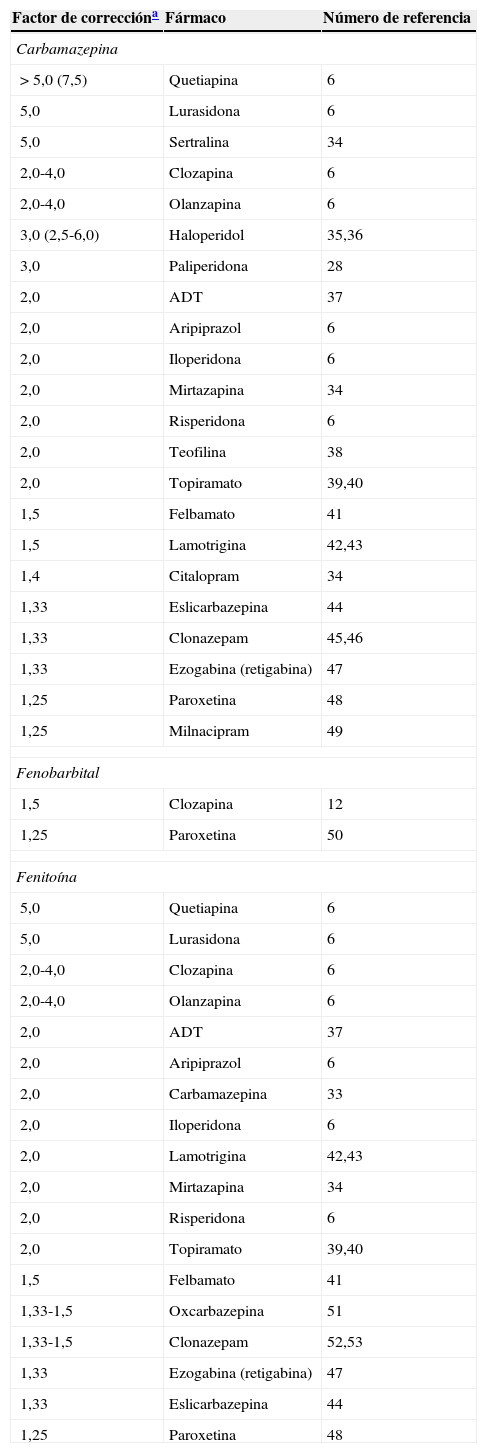
La tabla 4 incluye los factores de corrección calculados por el autor, utilizando la literatura disponible6,14,32,38–57. La administración de un inductor potente requiere el incremento de la dosis del sustrato. Por ejemplo, un factor de corrección de 2,0 indica que debe duplicarse la dosis del sustrato. Si el paciente está tomando un inductor y un sustrato, y se suspende el inductor para mantener la misma concentración plasmática del sustrato, se deberá utilizar el inverso del factor de corrección 1/2,0=0,5, o reducir la dosis a la mitad. La tabla 4 proporciona las recomendaciones basadas en la limitación de la información disponible; a medida que se publiquen más artículos, deberán recalcularse dichos factores de corrección. Se incluyen referencias para aquellos casos en los que el lector desee actualizar las mismas. Recuerden que los factores de corrección constituyen correcciones medias para los pacientes promedio; por tanto, una persona puede tener una capacidad metabólica superior o inferior a la media. A veces puede utilizarse la MT para realizar correcciones más precisas en una persona en particular6,25.
Factores de corrección para inductores potentes
| Factor de correccióna | Fármaco | Número de referencia |
|---|---|---|
| Carbamazepina | ||
| > 5,0 (7,5) | Quetiapina | 6 |
| 5,0 | Lurasidona | 6 |
| 5,0 | Sertralina | 34 |
| 2,0-4,0 | Clozapina | 6 |
| 2,0-4,0 | Olanzapina | 6 |
| 3,0 (2,5-6,0) | Haloperidol | 35,36 |
| 3,0 | Paliperidona | 28 |
| 2,0 | ADT | 37 |
| 2,0 | Aripiprazol | 6 |
| 2,0 | Iloperidona | 6 |
| 2,0 | Mirtazapina | 34 |
| 2,0 | Risperidona | 6 |
| 2,0 | Teofilina | 38 |
| 2,0 | Topiramato | 39,40 |
| 1,5 | Felbamato | 41 |
| 1,5 | Lamotrigina | 42,43 |
| 1,4 | Citalopram | 34 |
| 1,33 | Eslicarbazepina | 44 |
| 1,33 | Clonazepam | 45,46 |
| 1,33 | Ezogabina (retigabina) | 47 |
| 1,25 | Paroxetina | 48 |
| 1,25 | Milnacipram | 49 |
| Fenobarbital | ||
| 1,5 | Clozapina | 12 |
| 1,25 | Paroxetina | 50 |
| Fenitoína | ||
| 5,0 | Quetiapina | 6 |
| 5,0 | Lurasidona | 6 |
| 2,0-4,0 | Clozapina | 6 |
| 2,0-4,0 | Olanzapina | 6 |
| 2,0 | ADT | 37 |
| 2,0 | Aripiprazol | 6 |
| 2,0 | Carbamazepina | 33 |
| 2,0 | Iloperidona | 6 |
| 2,0 | Lamotrigina | 42,43 |
| 2,0 | Mirtazapina | 34 |
| 2,0 | Risperidona | 6 |
| 2,0 | Topiramato | 39,40 |
| 1,5 | Felbamato | 41 |
| 1,33-1,5 | Oxcarbazepina | 51 |
| 1,33-1,5 | Clonazepam | 52,53 |
| 1,33 | Ezogabina (retigabina) | 47 |
| 1,33 | Eslicarbazepina | 44 |
| 1,25 | Paroxetina | 48 |
ADT: antidepresivo tricíclico.
El factor de corrección del bupropión para la carbamazepina fue de 10,0, calculado por el autor a partir de la limitada información disponible49.
La carbamazepina es un FAE clásico aprobado en Estados Unidos para la epilepsia de inicio parcial, el trastorno bipolar y la neuralgia del trigémino20. La carbamazepina es metabolizada principalmente por el CYP3A (con una contribución menor del CYP2C8) a su metabolito activo, 10,11-epóxido, que puede representar el 40% del metabolismo de la carbamazepina, aunque la proporción es incluso mayor en pacientes con actividad del CYP3A4 inducida. El epóxido es transformado seguidamente en el diol inactivo por la enzima epóxido hidrolasa. Otras rutas incluyen la hidroxilación aromática (25%), posiblemente por el CYP1A2, y la glucuronidación de la cadena lateral de carbamoil por las UGT, principalmente por la UGT2B720.
La carbamazepina induce su propio metabolismo (tabla 2), que se triplica20. Al iniciar el tratamiento con carbamazepina, los niveles pueden no situarse en estado estacionario durante las primeras 3 a 5 semanas, debido al incremento progresivo de la autoinducción. La tabla 4 incluye los factores de corrección para diversos fármacos durante el tratamiento con la carbamazepina.
FenobarbitalEl fenobarbital es un FAE clásico aprobado en Estados Unidos para la epilepsia generalizada y la de inicio parcial, y para sedación20. Parte del fenobarbital se elimina sin alterar en la orina (20-50%) y el resto es metabolizado a parahidroxifenobarbital y fenobarbital N-glucósido. El CYP2C9 tiene un papel principal en la formación del parahidroxifenobarbital. Otras enzimas metabolizadoras menores son el CYP2C19 y el CYP2E120.
La literatura no describe que el fenobarbital sufra autoinducción (tabla 2). Conociendo su perfil inductivo de ser un inductor del CYP2C9, es posible que se produzca autoinducción, pero que sea probablemente modesta y no fácil de detectar, debido a la extraordinariamente larga vida media del fenobarbital (varios días). La tabla 4 incluye los factores de corrección únicamente para la clozapina y la paroxetina, ya que no existe información para otros fármacos. En caso de necesidad, se recomienda utilizar los factores de corrección para la carbamazepina y/o fenitoína como aproximaciones.
La literatura sugiere que los efectos inductivos del fenobarbital son leves (y probablemente sin relevancia clínica, <1/3 de descenso de sus niveles en sangre) para diversos FAE que incluyen: felbamato, lacosamida, levetiracetam, pregabalina y rufinamida20.
FenitoínaLa fenitoína es un FAE clásico aprobado en Estados Unidos para la epilepsia generalizada y la de inicio parcial, y la prevención de las crisis epilépticas secundarias a la cirugía craneal y los traumatismos craneales20. La fenitoína es ampliamente para-hidroxilada por el CYP2C9, mientras que el CYP2C19 puede constituir la segunda enzima más importante para este paso. Otras enzimas menos importantes pueden ser el CYP2C8 y el CYP3A20. La fenitoína es un leve inductor de su propio metabolismo, con arreglo a un estudio en voluntarios58, que es compatible con estudios in vitro que reflejan efectos inductivos únicamente leves o moderados en la subfamilia CYP2C35. La autoinhibición es más importante que la autoinducción. Los clínicos deben saber que la fenitoína puede inhibir su metabolismo mediante la saturación de CYP2C9 y CYP2C19. La fenitoína puede describirse farmacológicamente como poseedora de una ventana terapéutica estrecha y con una farmacocinética no lineal. Su cinética depende de la dosis, y su capacidad es limitada. Según la experiencia del autor, una dosificación mayor al rango superior de la ventana terapéutica (>20μg/ml) produce la saturación de las enzimas y el incremento de la vida media. Esto requiere la suspensión completa del tratamiento con fenitoína durante un mínimo de 2 o 3 días, hasta que el metabolismo se normalice y recupere los niveles de <20μg/ml, y usar MT repetida hasta que se normalicen las concentraciones. En situaciones de elevada concentración plasmática de fenitoína (cercana a 20μg/ml), la administración de cualquier inhibidor de los CYP2C, o de cualquier fármaco que compita con el CYP2C9 y/o el CYP219, puede verse acompañada de incrementos considerables de la concentración que no se producirían probablemente si las concentraciones de fenitoína fueran sensiblemente menores (p.ej., 10μg/ml). La tabla 4 incluye los factores de corrección para diversos fármacos durante el tratamiento con la fenitoína.
PrimidonaLa primidona es un FAE clásico, aprobado en Estados Unidos para la epilepsia generalizada y la de inicio parcial. Se utiliza raramente solo pacientes epilépticos resistentes a otros tratamientos, y en aquellos con temblor esencial20. Hasta un cuarto de la primidona se elimina sin alterar en la orina, y el resto es metabolizado por las isoenzimas CYP2C a 2 metabolitos activos: fenobarbital y feniletilmalonamida. Como la primidona se transforma en fenobarbital, debería considerarse como un inductor tan potente como este último20. La tabla 4 no incluye factores de corrección para la primidona; en caso de necesidad, el autor sugeriría el uso de los factores de corrección para la fenitoína, como aproximación.
Efecto de los inductores potentes en los grandes grupos de fármacosEsta subsección realiza comentarios sobre los FAE y los fármacos psiquiátricos y médicos. El metabolismo de los FAE es demasiado heterogéneo para revisarse brevemente como grupo, pero la tabla 4 aporta datos sobre los factores de corrección para diversos de ellos, cuando se administran FAE potentemente inductores. En esta sección se revisan brevemente 3 grupos de fármacos psiquiátricos (antipsicóticos, antidepresivos y benzodiazepinas), así como fármacos no neuropsicofarmacológicos metabolizados por el CYP3A4.
Cuando los clínicos consideran la administración de un potente inductor a un paciente que toma antipsicóticos, deberían valorar el riesgo de la inducción y revisar la literatura más reciente. Una revisión reciente de los antipsicóticos de segunda generación6 indicaba que estos fármacos pueden dividirse en 3 grupos: los que necesitan un incremento de dosis masivo, los que necesitan un incremento mínimo o nulo, y los del grupo intermedio, que necesitan un incremento de dosis moderado. La lurasidona y la quetiapina precisan incrementos de dosificación masivos (tabla 4). En dicho artículo de revisión de 2012, la amisulprida, la paliperidona y la ziprasidona fueron clasificadas como de incremento mínimo o nulo, pero, como se ha indicado previamente, el reciente estudio de Yasui-Furukori et al.32 ha demostrado que debería eliminarse de este grupo de antipsicóticos a la paliperidona, ya que su incremento es moderado tras la administración de inductores potentes de los FAE. Por ello, los antipsicóticos de segunda generación con un potencial intermedio de inducción son los que dependen del CYP1A2 y las UGT (clozapina y olanzapina), los que dependen parcialmente del CYP3A4 (aripiprazol, iloperidona y risperidona), y probablemente la paliperidona (tabla 4). Actualmente, el autor no tiene información para indicar si los inductores potentes influyen o no en el metabolismo de la asenapina6. Existe información muy limitada acerca de los factores de corrección para los antipsicóticos de primera generación (véase el haloperidol en la tabla 4).
La información sobre los efectos de los inductores potentes en los antidepresivos tricíclicos (ADT) es limitada. Las aminas terciarias tales como la amitriptilina, la clomipramina y la imipramina son desmetiladas por el CYP2C19 y, en menor medida, por el CYP1A2, el CYP2C9 y el CYP3A4; todos estos CYP pueden ser inducidos10. Las aminas secundarias, tales como la nortriptilina y la desimipramina, son hidroxiladas por el CYP2D6 y posiblemente por otros CYP10. Por tanto, si un paciente toma un ADT a la vez que un inductor potente de los ADT, deberá realizarse MT. La tabla 4 proporciona un factor de corrección aproximado de 2,0, aunque el rango, descrito en esa revisión41, es de 1,4-2,5. Esto indica que es mejor realizar MT del ADT al tratar de corregir el efecto de un inductor potente en un paciente al que se administra un ADT. Existe información limitada acerca de los efectos de los inductores potentes en el metabolismo de los antidepresivos de segunda generación8,38,53,59. La tabla 4 incluye los factores de corrección de citalopram, milnacipram, mirtazapina, sertralina y paroxetina en pacientes que toman carbamazepina, de paroxetina en pacientes que toman fenobarbital, y de citalopram y paroxetina en pacientes que toman fenitoína. El bupropión se incluye como nota al pie en la tabla 4, ya que el factor de corrección calculado por el autor a partir de la limitación de información disponible es de 10,08. El bupropión es metabolizado principalmente por el CYP2B6, que es muy sensible a la inducción; por tanto, debería evitarse el bupropión en pacientes a quienes se administren inductores potentes, ya que la farmacia puede no querer dispensar bupropión en dosis 10 veces superiores a lo normal. La reboxetina y la vilazodona, que son metabolizadas principalmente por el CYP3A4, pueden ser particularmente sensibles a la inducción. De acuerdo con la información de los antipsicóticos dependientes del CYP3A4, estos fármacos pueden precisar un factor de corrección de al menos 58.
El metabolismo de las benzodiazepinas es heterogéneo, aunque existen 3 grandes grupos: los metabolizados por: a)el CYP3A4; b)el CYP2C19 y el CYP3A4, y c)las UGT. El CYP3A4 explica el 93%60 del metabolismo del triazolam, el 92%60 del midazolam y el 75%60 del alprazolam. Es probable que estas 3 benzodiazepinas sean fuertemente inducidas por los inductores potentes, debiendo evitarse su uso en pacientes que toman dichos inductores. El clonazepam es parcialmente metabolizado por el CYP3A4. La tabla 4 incluye sus factores de corrección para la carbamazepina y la fenitoína. El lorazepam y el oxazepam no son metabolizados por los CYP, sino por las UGT61. No existen estudios, pero es probable que los inductores potentes tengan efectos clínicamente relevantes sobre el lorazepam y el oxazepam. De igual modo, no existen buenos estudios acerca de los efectos de los inductores potentes sobre el diazepam y el clobazam, que son metabolizados por el CYP3A4 y el CYP2C19. Los limitados datos sobre el clobazam62,63 indican que, aunque es probable que la carbamazepina sea un inductor, la fenitoína puede inducir el CYP3A4 y también competir con el metabolismo del clobazan a traves del CYP2C196.
Los 3 FAE son potentes inductores del CYP3A4. Muchos fármacos no neuropsicofarmacológicos son ampliamente metabolizados por el CYP3A4, incluyendo muchos de los bloqueadores del canal del calcio, las estatinas, los inmunosupresores y los anticonceptivos orales con estrógenos. Los clínicos que combinan la carbamazepina, el fenitoína o el fenobarbital con cualquier fármaco de estos tipos debe prestar atención a la falta de eficacia asociada a la disminución de las concentraciones plasmáticas, y considerar el incremento de las dosis de estos sustratos, o intercambiarlos por fármacos similares no metabolizados por el CYP3A4. Si los fármacos son metabolizados principalmente por el CYP3A4, el factor de corrección será ≥5. Esto se basa en el factor de corrección de la quetiapina (tabla 4), que es metabolizada fundamentalmente (85%) por el CYP3A464. El caso de los agentes antirretrovirales es particularmente complejo. Deberán evitarse los 3 FAE que son potentes inductores en pacientes que toman inhibidores de la proteasa o inhibidores no nucleósidos de la transcriptasa inversa65.
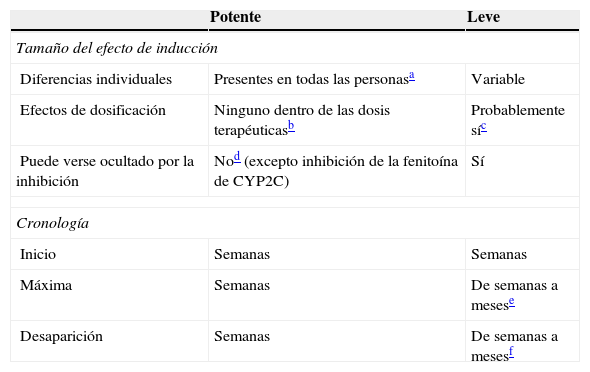
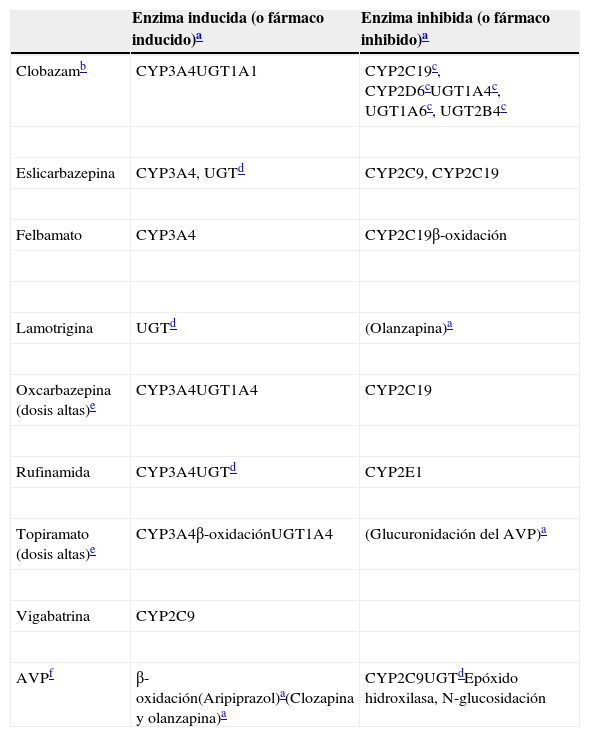
Inductores levesEste artículo clasifica a la clobazam, la eslicarbazepina, el felbamato, la lamotrigina, la oxcarbazepina, la rufinamida, el topiramato, la vigabatrina y el AVP como inductores leves. En la medida del conocimiento del autor, ningún otro autor ha agrupado del mismo modo a estos fármacos, ni los ha contrastado con los inductores potentes (tabla 5). Por ello, el resumen de sus enzimas inductivas incluye sus propiedades inhibitorias (tabla 6), para ayudar al lector a comprender que estos FAE tienen un perfil de interacción farmacológica muy complejo.
Inductores leves: comparación con los inductores potentes
| Potente | Leve | |
|---|---|---|
| Tamaño del efecto de inducción | ||
| Diferencias individuales | Presentes en todas las personasa | Variable |
| Efectos de dosificación | Ninguno dentro de las dosis terapéuticasb | Probablemente síc |
| Puede verse ocultado por la inhibición | Nod (excepto inhibición de la fenitoína de CYP2C) | Sí |
| Cronología | ||
| Inicio | Semanas | Semanas |
| Máxima | Semanas | De semanas a mesese |
| Desaparición | Semanas | De semanas a mesesf |
Aunque no ha sido estudiado sistemáticamente, se acepta generalmente que los inductores potentes tienden a inducir máximamente a todos los pacientes, siempre que se les hayan administrado dosis típicas que se considera que causan una inducción máxima.
Se piensa también que una dosis terapéutica para la epilepsia debería causar una inducción máxima en todos los pacientes. Por tanto, el incremento adicional de las dosis superior a las dosis terapéuticas puede no causar más inducción. De igual modo, la administración de otro inductor potente a una persona que toma dosis normales de un inductor potente puede no suponer diferencia alguna.
Consúltese en el texto la información sobre los efectos de la dosificación de la oxcarbazepina, el topiramato y el AVP.
Inductores leves: propiedades inductivas e inhibitorias
| Enzima inducida (o fármaco inducido)a | Enzima inhibida (o fármaco inhibido)a | |
|---|---|---|
| Clobazamb | CYP3A4UGT1A1 | CYP2C19c, CYP2D6cUGT1A4c, UGT1A6c, UGT2B4c |
| Eslicarbazepina | CYP3A4, UGTd | CYP2C9, CYP2C19 |
| Felbamato | CYP3A4 | CYP2C19β-oxidación |
| Lamotrigina | UGTd | (Olanzapina)a |
| Oxcarbazepina (dosis altas)e | CYP3A4UGT1A4 | CYP2C19 |
| Rufinamida | CYP3A4UGTd | CYP2E1 |
| Topiramato (dosis altas)e | CYP3A4β-oxidaciónUGT1A4 | (Glucuronidación del AVP)a |
| Vigabatrina | CYP2C9 | |
| AVPf | β-oxidación(Aripiprazol)a(Clozapina y olanzapina)a | CYP2C9UGTdEpóxido hidroxilasa, N-glucosidación |
AVP: ácido valproico.
Para los fármacos entre paréntesis no se ha establecido con carácter definitivo la enzima tras la inducción o la inhibición.
En los años setenta, el clobazam fue aprobado primeramente en Australia y a continuación en Francia para su uso en casos de ansiedad y epilepsia. En 2011 fue aprobado en Estados Unidos para el tratamiento adyuvante de crisis epilépticas asociadas al síndrome de Lennox-Gastaut9. La principal ruta metabólica del clobazam conlleva la N-desmetilación, principalmente por el CYP3A4, y en menor medida por el CYP2C19 y el CYP2B6. El N-desmetilclobazam es un metabolito activo a su vez metabolizado, principalmente por el CYP2C19. El N-desmetilclobazam y sus metabolitos incluyen aproximadamente el 94% de los compuestos de la orina producidos por este fármaco9.
Los estudios in vitro indican62,63 que: a)el N-desmetilclobazam es un inhibidor débil del CYP2D6, el CYP2C9, la UGT1A4, la UGT1A6 y la UGT2B4, y b)el clobazam y el N-desmetilclobazam inducen la actividad del CYP3A4 de un modo que depende de la concentración. Es sorprendente que el clobazam sea un inductor leve del CYP3A4, ya que el clobazam es una benzodiazepina, y estas no están consideradas como inductores.
Por tanto, el clobazam autoinduce su propio metabolismo (ya que es metabolizado por el CYP3A4 y es un inductor del CYP3A4); sin embargo, al revisar atentamente un antiguo estudio66, este autor1 ha concluido que la autoinducción puede no producirse hasta la tercera semana del tratamiento. Según indican los estudios in vitro, los efectos inductivos dependen tanto del clobazam como del N-desmetilclobazam, y dependen de sus concentraciones. Es importante saber que el N-desmetilclobazam precisa diversas semanas para alcanzar el estado estacionario9, y que es probable que la máxima inducción del clobazam precise de varios meses hasta que el N-desmetilclobazam alcance completamente un estado estacionario.
El clobazam constituye definitivamente un inductor del midazolam63, lo que no es sorprendente dado que este último es principalmente metabolizado (92%)60 por el CYP3A4. El prospecto de Estados Unidos ofrece información conflictiva acerca de la relevancia clínica de la inducción del clobazam, ya que propone que no se precisa ajustar la dosificación de los fármacos que están metabolizados principalmente por el CYP3A4, pero luego describe que la administración de clobazam puede estar asociada a la pérdida de eficacia de los anticonceptivos orales62, que debería explicarse por la inducción del CYP3A4.
La literatura aporta información conflictiva sobre los efectos inductivos del clobazam en la carbamazepina, que es principalmente metabolizada por el CYP3A4, pero al mismo tiempo induce su propio metabolismo y puede ser parcialmente inhibida por el N-desmetilclobazam1. De igual modo, la información sobre el clobazam, acerca de la posibilidad de inducción de las UGT, es conflictiva1. La información sobre prescripción del clobazam en Estados Unidos62 reportaba que los estudios farmacocinéticos en la población durante los ECA con clobazam no indicaban efectos sobre el metabolismo del AVP y la lamotrigina, mientras que algunos estudios de MT67, aunque no todos1, indicaban que el clobazam puede ser un inductor leve del metabolismo de la lamotrigina. Un estudio de MT describió que el clobazam puede reducir levemente las concentraciones séricas de levetiracetam68.
EslicarbazepinaLa eslicarbazepina acetato es un FAE de segunda generación lanzado al mercado europeo en 2009 para el tratamiento adyuvante en pacientes adultos con epilepsia de inicio parcial69, aunque no está disponible aún en Estados Unidos. Tras su absorción, la eslicarbazepina acetato se hidroliza (95%) rápida y ampliamente en el metabolismo de primer paso en hígado e intestino a eslicarbazepina (también conocida como S-licarbazepina o S-MHD). Otro 5% se oxida a oxcarbazepina y R-licarbazepina (o R-MHD). Los metabolitos eliminados en la orina incluyen 2/3 de la dosis total como S-MHD y 1/3 como conjugado de glucuronida, probablemente por la mediación de la UGT1A170. La eslicarbazepina acetato es principalmente un profármaco de S-MHD69.
La eslicarbazepina no autoinduce su propio metabolismo, pero parece ser un inductor clínicamente relevante del CYP3A4, ya que causa un aumento de la eliminación de simvastatina del doble71, y del etinilo estradiol y levonorgestrel presentes en los anticonceptivos orales, de manera dependiente de la dosis72. La eslicarbazepina puede ser también un inductor leve de las UGT, incrementando ligeramente (<20%) la eliminación de diversos FAE que incluyen: la carbamazepina, la lamotrigina y el topiramato48. Los estudios in vitro indican que la eslicarbazepina es un inhibidor moderado del CYP2C9 y del CYP2C19, lo cual es compatible con el aumento de un tercio en los niveles de fenitoína48.
FelbamatoEl felbamato es un FAE de segunda generación aprobado en Estados Unidos para la epilepsia, que responde inadecuadamente a los tratamientos alternativos, y que es tan grave que el riesgo sustancial de anemia aplástica y/o fallo hepático es presuntamente aceptable20. Aproximadamente el 40-60% del felbamato es excretado por los riñones y es metabolizado por hidroxilación (por el CYP3A y el CYP2E1) y glucuronidación. El CYP3A contribuye en gran medida a su metabolismo cuando se prescriben inductores. El felbamato es un inhibidor del CYP2C19 y de la β-oxidación, y es un inductor del CYP3A20. Existe información limitada sobre la relevancia de los efectos inductivos del felbamato.
LamotriginaLa lamotrigina es un FAE de segunda generación aprobado en Estados Unidos para diversos tipos de epilepsia, y como tratamiento de mantenimiento en el trastorno bipolar20. La glucuronidación es la ruta metabólica principal, y representa hasta el 65-90% del metabolismo de la lamotrigina61. El metabolito principal es la 2-N-glucuronida que es inactiva. La lamotrigina y sus metabolitos se eliminan en la orina. Las revisiones describen que la UGT1A4 metaboliza la lamotrigina. Existe desacuerdo acerca de la importancia de la UGT27B como enzima metabólica de la lamotrigina20.
La lamotrigina es un inductor leve de la glucuronidación y de su propio metabolismo (<20% de reducción en 2semanas). No se observa el efecto de esta autoinducción en pacientes a los que ya se administran inductores más potentes20.
La lamotrigina puede estar asociada a pequeñas (<25%) reducciones de los niveles del AVP73. Existen relativamente pocos estudios bien controlados sobre los efectos de la lamotrigina en los niveles antipsicóticos5,6. En un estudio de MT sobre la quetiapina74 se describió una disminución leve del metabolismo de la quetiapina, pero en otro estudio de MT más reciente se ha descrito una disminución de aproximadamente la mitad, lo que requerirá la duplicación de la dosis de quetiapina75. Los mejores estudios sobre clozapina no reflejan grandes efectos sobre los niveles de clozapina5,6. La información sobre olanzapina es más complicada, e incluye un estudio in vitro que sugiere que las grandes concentraciones de lamotrigina pueden inhibir el metabolismo de la olanzapina76. Los estudios clínicos no han demostrado los efectos inhibitorios de la lamotrigina5,6. La falta de consideración otorgada al estatus de fumador puede ser una explicación; la lamotrigina puede comportarse como un inhibidor leve del metabolismo de la olanzapina únicamente en fumadores15. No es de esperar que la lamotrigina influya sobre los antidepresivos de segunda generación8,38.
OxcarbazepinaLa oxcarbazepina es un FAE de segunda generación aprobado en Estados Unidos para la epilepsia de inicio parcial20. Algunos clínicos, basándose en su similitud con la estructura química de la carbamazepina, utilizan la oxcarbazepina para el trastorno bipolar y la neuralgia del trigémino.
La oxcarbazepina es rápidamente reducida por la arilcetona reductasa citoplásmica a MHD, denominada también licarbazepina, que es el metabolito clínicamente relevante20. Por tanto, la oxcarbazepina acetato es un profármaco de ambos enantiómeros S-MHD (80%) y R-MHD (20%)69. El MHD se elimina por glucuronidación y, en menor medida, por oxidación a un metabolito inactivo. La excreción renal es la principal ruta de excreción de la oxcarbazepina (80% de la dosis), incluyendo los glucurónidos del MHD (40%), el MHD inalterado (27%), los conjugados de MHD u oxcarbazepina (13%), y en menor proporción el 10,11-dihidroximetabolito inactivo20.
La oxcarbazepina no es inductora de su propio metabolismo, a diferencia de carbamazepina que lo es77. La oxcarbazepina es un inductor débil del CYP3A y las enzimas de glucuronidación, y un débil inhibidor del CYP2C19.
En general, la literatura está de acuerdo que sus efectos inductivos son menos pronunciados que los de la carbamazepina78. Existe más desacuerdo acerca de la intensidad de los efectos inductivos de la oxcarbazepina y si estos deben ignorarse o son clínicamente relevantes. Lamentablemente, la literatura no ha prestado atención a Patsalos et al.79, quienes propusieron que únicamente las dosis de oxcarbazepina ≥1.500mg/día podrían tener efectos inductivos, ya que esto podría explicar los hallazgos conflictivos. La oxcarbazepina normalmente no muestra efectos inductivos en las dosis bajas o moderadas utilizadas en estudios controlados, aunque parece un inductor leve pero clínicamente relevante en los estudios naturalistas. En el mejor estudio farmacocinético publicado, Andreasen et al.80 compararon 17 días de administración de 1.200mg/día y 800mg/día de carbamazepina en voluntarios sanos; la eliminación del metabolito quinidina dependiente de CYP3A experimentó un incremento respectivo de cerca del 90 y del 180%. Si asumimos que este estudio refleja con precisión lo que les sucede a los fármacos metabolizados por el CYP3A4, deberían duplicarse al menos las dosis de estos fármacos al administrarse la oxcarbazepina, y casi triplicarse al administrarse la carbamazepina.
Un estudio controlado que utilizó 900-1.200mg/día durante 5semanas no reflejó cambios significativos en el metabolismo de la risperidona o la olanzapina81. Los clínicos deberán ser conscientes de que la oxcarbazepina, particularmente en dosis elevadas, puede incrementar el metabolismo de algunos fármacos psiquiátricos. De hecho, se ha publicado un caso clínicamente relevante de descenso de los niveles de clomipramina tras la administración de la oxcarbazepina82.
Un estudio controlado en sujetos sanos, realizado por una empresa farmacéutica, sugirió que las dosis de oxcarbazepina de 1.200mg/día no influían sobre el metabolismo de la lamotrigina en pacientes con tomas de 200mg/día83. Un estudio de MT que incluía tratamientos a largo plazo sugirió que los niveles séricos de lamotrigina pueden reducirse levemente al administrar conjuntamente oxcarbazepina, debiendo incrementar las dosis de lamotrigina por cerca del 20-30%47,84. La relevancia clínica de esta interacción farmacológica viene demostrada por 2 casos de úlceras orales inducidas por lamotrigina (signos iniciales del síndrome de Stevens-Johnson) a los 2meses después de haberse suspendido la administración de la oxcarbazepina12. La oxcarbazepina reduce los niveles del levetiracetam y el topiramato en <30%78,85. La oxcarbazepina puede ser también un inductor menor (21% de disminución) de las carboxilesterasas que metabolizan la rufinamida86.
En resumen, las dosis elevadas de oxcarbazepina pueden inducir el CYP3A4, la UGT1A4 y posiblemente otras enzimas metabólicas. Se necesitan estudios controlados utilizando dosis de oxcarbazepina ≥1.200mg/día para establecer la magnitud de sus efectos inductivos en el paciente promedio y el factor de corrección necesario para compensar dichos efectos. Los estudios naturalistas y los reportes de casos deben considerar la posibilidad de que algunas personas sean particularmente sensibles a los efectos inductivos de la oxcarbazepina, y demostrar la inducción en dosis menores. La desaparición de la la inducción por oxcarbazepina puede emplear hasta 2meses en manifestarse completamente.
RufinamidaLa rufinamida es un FAE de segunda generación aprobado en Estados Unidos únicamente para su uso en el síndrome de Lennox-Gastaut20. La rufinamida se metaboliza ampliamente (solo el 2-4% se excreta inalterado en orina y heces). No es metabolizada por los CYP sino por las carboxilesterasas. No existen metabolitos activos conocidos20. La rufinamida es un inhibidor leve del CYP2E1, un inductor leve del CYP3A, y posiblemente un inductor leve de algunas enzimas de la glucuronidación20.
Los efectos de la rufinamida sobre otros niveles de antiepilépticos pueden no tener relevancia clínica. Parecen producirse descensos leves (<15%) de los niveles de carbamazepina y lamotrigina87. Actualmente, otra interacción farmacológica de la rufinamida que se considera relevante es que este fármaco es un inductor de los anticonceptivos orales. La rufinamida puede no tener efectos sobre los niveles de topiramato y AVP87.
Los niveles plasmáticos de los fármacos psiquiátricos metabolizados por CYP3A pueden reducirse levemente con la administración conjunta de rufinamida, debido a la inducción de CYP3A, aunque esta interacción no ha sido estudiada sistemáticamente excepto para el triazolam, un sustrato de CYP3A4 (que puede representar el 93%60 del metabolismo del mismo). La rufinamida incrementó el metabolismo del triazolam probablemente en un tercio, aunque esto se estimó con una única dosis de triazolam87. Es difícil extrapolarlo a la dosificación repetida de los sustratos de CYP3A4 en la práctica clínica, ya que la rufinamida podría tener una influencia mucho mayor.
TopiramatoEl topiramato es un FAE de segunda generación aprobado en Estados Unidos para diversos tipos de epilepsia y profilaxis de la migraña20, y combinado con fentermina, para la pérdida de peso.
El topiramato se elimina inalterado principalmente en la orina, aunque es parcialmente metabolizado por los CYP (aproximadamente el 20%). La importancia relativa del metabolismo por los CYP se incrementa al administrar un inductor potente tal como la fenitoína o la carbamazepina, que duplica la eliminación del topiramato, debido a un incremento del metabolismo por los CYP29.
El topiramato se muestra también como un inductor débil de diversas enzimas metabólicas, aunque la inducción puede verse influida por las dosis de este fármaco. Tras un estudio in vitro, Nallani et al.88 propusieron que en dosis ≥400mg/día, las propiedades inductivas del topiramato pueden tener una significación clínica para los sustratos del CYP3A. En un grupo de 12 mujeres epilépticas tratadas con dosis estables de AVP, junto con una combinación de noretindrona, 1mg/etinilestradiol, comprimido de 35μg, dosis de topiramato de 200mg/día, 400mg/día, u 800mg/día, se originó un descenso estadísticamente significativo y de la media de la AUC del etinilestradiol de una manigtud del 18 al 30% y que aumenta con la dosis desde 200 a 800 mg/dia.89. La farmacocinética de la noretindrona permaneció intacta. Como contraste, otro estudio encontró que las dosis de topiramato de 50 a 200mg/día no afectaban significativamente a la eliminación del etinilestradiol o la noretindrona90.
El topiramato puede tener efectos complejos sobre el AVP, puesto que puede actuar como inductor al incrementar la β-oxidación, aunque también puede inhibir la glucuronidación del AVP. A bajas dosis de AVP, la β-oxidación es la ruta metabólica más importante de dicho fármaco, y el topiramato puede comportarse como un inductor del metabolismo del mismo91. A dosis elevadas de valproato, la glucuronidación es la ruta metabólica más importante de dicho fármaco, y el topiramato puede comportarse como un inhibidor del metabolismo del mismo92.
Un pequeño estudio controlado realizado por la empresa farmacéutica, con incrementos progresivos de las dosis de hasta 400mg/día durante 2semanas, no reflejó ninguna interacción farmacológica relevante entre la lamotrigina y el topiramato93. En un estudio controlado realizado por investigadores independientes se añadió de 100 mg/día a 800 mg/día de topiramato según se toleraba al tratamiento mensual de lamotrigina. Este estudio demostró que en 4 de los 7 pacientes se produjo una disminución del metabolismo de la lamotrigina del 40-50%94. Reimers et al.95, en un estudio grande de MT controlando por factores de confusión, describieron que el topiramato puede disminuir los niveles de lamotrigina, pero no se describieron las dosis de topiramato.
En resumen, las dosis elevadas de topiramato pueden inducir el CYP3A4 y algunas otras enzimas (tabla 6). Son necesarios estudios controlados de topiramato con dosis ≥400mg/día para establecer la magnitud de sus efectos inductivos en los pacientes promedio, así como los factores de corrección necesarios para compensar su efecto. Los estudios naturalistas y los reportes de casos deben considerar la posibilidad de que algunas personas puedan ser particularmente sensibles a los efectos inductivos del topiramato, y demostrarlos en dosis menores. Un factor de complicación es que en algunas situaciones el topiramato puede tener propiedades inhibitorias del metabolismo de los fármacos.
VigabatrinaLa vigabatrina fue inicialmente aprobada en Gran Bretaña y la República de Irlanda en 1989 como agente antiepiléptico. A finales de los noventa había sido aceptado en la práctica clínica convencional para el cuidado de pacientes adultos y pediátricos, aunque se reportaron defectos severos y persistentes en el campo visual asociados a la vigabatrina. Veinte años después se introdujo la vigabatrina en el mercado de Estados Unidos para pacientes con crisis epilépticas parciales, complejas y refractarias que no respondan adecuadamente a diversos tratamientos alternativos, y en los que los beneficios potenciales sobrepasaban el riesgo de la pérdida de visión.
De acuerdo al prospecto en Estados Unidos96, la vigabatrina no se metaboliza de modo significativo y es eliminada principalmente mediante excreción renal. En un estudio de isótopos, utilizando una dosis única de vigabatrina para explorar su eliminación, Durham et al.97 hallaron que cerca del 95% de la radiactividad total se recuperaba en la orina, y que el fármaco madre representaba la mayoría de la radiactividad (82%); uno de los metabolitos representaba el 3-5% y el otro, el 1-2%. No existen estudios si que la repetición de las dosis incrementa, o no, la fracción metabolizada de la vigabatrina. Incluso suponiendo que <10% de la vigabatrina se metabolice, no hay duda de que la fracción de la vigabatrina que lo hace se incrementa notablemente en los pacientes que toman inductores, aunque dicho incremento no ha sido lo suficientemente cuantificado en la literatura1. La vigabatrina se ha prescrito raramente como monoterapia, y la mayoría de los pacientes estudiados en la literatura se les administraban conjuntamente otros FAE, sobre todo inductores potentes tales como la carbamazepina o fenitoína. Por tanto, en el paciente típico de vigabatrina, el metabolismo puede ser clínicamente relevante e incluir más del 10% de la dosis, debido a la presencia de inductores.
Un estudio in vitro utilizado para obtener la aprobación para el mercado de Estados Unidos demostró que la vigabatrina es un inductor de CYP2C996, lo que explica algunos estudios antiguos describiendo la reducción de las concentraciones plasmáticas de fenitoína del 25%, que no eran evidentes hasta la 5.a o la 6.a semana98. El prospecto en Estados Unidos96 recomienda que, aunque no es preciso realizar ajustes rutinarios de la dosificación de fenitoína, deberán considerarse dichos ajustes de indicarse clínicamente. También se han descrito en pacientes con vigabatrina descensos leves de los niveles del AVP y del fenobarbital96.
Ácido valproicoEl AVP es un FAE clásico que fue aprobado en Estados Unidos para tratar diversos tipos de epilepsia, trastorno bipolar o profilaxis de la migraña20. La mayoría del AVP se metaboliza en el hígado, eliminándose <5% inalterado en la orina. Las dos vías principales de matabolismo son las UGT (40%) y la β-oxidación como ácido graso (30%), mientras que los CYP (incluyendo el CYP2C9, el CYP2C19 y el CYP2A6) son una vía menor. A dosis bajas la β-oxidación puede ser la ruta más importante, mientras que a dosis terapéuticas la glucuronidación puede tener más relevancia. Muchas UGT parecen estar implicadas en la glucuronidación del valproato, incluyendo la UGT1A3, la UGT1A4, la UGT1A6, la UGT1A9 y la UGT2B7, así como la UGT1A8 y la UGT1A10 que están localizadas en el intestino20.
El AVP es considerado normalmente un inhibidor de diversas enzimas (tabla 6), incluyendo el CYP2C9, la epoxidohidroxilasa, diversas UGT y la ruta de la N-glucosidación del fenobarbital20. Los estudios en ratones parecen mostrar que el AVP autoinduce su propia glucuronidación99. La primera sugerencia sobre este hecho partió de otros estudios centrados en otros fármacos (en el felbamato100 y la lamotrigina101). La exploración de sus efectos sobre el AVP llevó a sus autores a cuestionarse si dicho ácido puede autoinducir su propio metabolismo en humanos.
Más recientemente, se ha acumulado información sobre sus propiedades inductivas. El AVP induce: a)su propio metabolismo, induciendo la β-oxidación (estudio prospectivo)102; b)la expresión genética del CYP3A4 y del P-gp (estudio in vitro)103; c)posiblemente la UGT1A1, hallada en un paciente que tomaba irinotecán (que tiene un metabolito activo el SN-38 metabolizado por UGT1A1)104; d)el metabolismo del aripiprazol en un grado leve (estudio prospectivo)105; e)el metabolismo de la olanzapina (serie de casos106, un estudio de MT107 y un estudio prospectivo de interacciones farmacológicas)15; f) el metabolismo de la clozapina (series de casos108,109, caso retrospectivo18 y un estudio con un modelo estadístico analizando muestras de MT y de estudios de interacciones farmacológicas14,19), y g)el metabolismo de la vitaminaD en un estudio in vitro110. La información disponible sobre el metabolismo de la clozapina-olanzapina indica que el AVP puede ser un inductor y/o un inhibidor competitivo. Su efecto neto (predominancia de inducción o inhibición) puede depender del tiempo, de la dosificación de AVP y de la presencia/ausencia de tabaco.
Otros efectos de los inductores de los fármacos antiepilépticos más allá de la interacción farmacológicaEn una excelente y reciente revisión de las propiedades inductivas de los FAE desde el punto de vista de la epilepsia, Brodie et al.111 aportan una perspectiva histórica reconociendo que el problema había sido estudiado durante los últimos 30años112, aunque la concienciación sobre su magnitud para el metabolismo endógeno se ha puesto de manifiesto solo en los últimos 10años. Las secciones previas se centraron en el metabolismo de los compuestos exógenos (denominados xenobióticos), pero los inductores potentes también tienen otras consecuencias sobre la salud a largo plazo, ya que sus propiedades inductivas pueden influir sobre el metabolismo de los compuestos endógenos y la homeostasis a diversos niveles, incluyendo a)las hormonas sexuales; b)la vitaminaD; c)las hormonas tiroideas; d)el metabolismo lipídico, y e)posiblemente el ácido fólico. En la experiencia del autor, probablemente debido a los mecanismos compensatorios endógenos, estos efectos inductivos de los compuestos endógenos tienen más probabilidad de detectarse y de tener una relevancia clínica cuando estos compuestos son administrados exógenamente.
Es evidente que los inductores del CYP3A4 tienen potentes efectos inductivos sobre las hormonas sexuales administradas exógenamente. Además, diversos inductores leves (clobazam, eslicarbazepina, oxcarbazepina, topiramato y rufinamida) han sido aprobados con advertencias sobre el riesgo de embarazo en mujeres que utilizan anticonceptivos orales como único método anticonceptivo. Los efectos inductivos se observan también cuando se administran hormonas sexuales para el tratamiento del hipopituitarismo113. El efecto de los inductores potentes sobre las hormonas sexuales masculinas y femeninas endógenas parece más limitado, aunque en casos raros puede asociarse a una disminución de la potencia sexual en varones y trastornos menstruales en mujeres114.
La vitaminaD, al igual que las hormonas sexuales, es un compuesto derivado del colesterol, por lo que los inductores potentes interfieren definitivamente en su metabolismo, que es mediado por las CYP, contribuyendo al riesgo de osteoporosis115. Además, los datos más recientes sugieren que los inductores leves, incluyendo la oxcarbazepina, el topiramato y el AVP, pueden originar también osteoporosis debido a la inducción del metabolismo de la vitaminaD110,116.
Las UGT son fundamentales para el metabolismo de las hormonas tiroideas, por lo que los inductores potentes pueden interferir en la función tiroidea117 al incrementar el metabolismo de dichas hormonas. Ocasionalmente, la oxcarbazepina se ha asociado también al hipotiroidismo113,117. Según la experiencia del autor, las anomalías tiroideas son más probables cuando la retroalimentación endógena no funciona, siendo frecuentes cuando la medicación tiroidea se administra de manera exógena y el cuerpo no puede compensar la leve disfunción tiroidea causada por los FAE. Aunque no se comprende bien, los inductores potentes pueden interferir en el metabolismo del colesterol y otros lípidos complejos, habiéndose asociado a la hiperlipidemia118.
Más controvertida es la idea de que los inductores potentes pueden influir en el metabolismo del ácido fólico, lo que puede contribuir a la hiperhomocisteinemia, un posible factor de riesgo de la aterosclerosis119,120. La fenitoína se asocia claramente al sobrecrecimiento gingival121; la fenitoína y el fenobarbital pueden causar anemia macrocítica122, que se ha asociado a las alteraciones metabólicas del ácido fólico. La literatura usualmente describe que la fenitoína puede ser un inhibidor del transportador del ácido fólico121,122.
En resumen, está bien establecido el efecto de los inductores potentes en el metabolismo endógeno de: a)las hormonas sexuales; b)la vitaminaD; c)las hormonas tiroideas; d)el metabolismo lipídico, y e)el ácido fólico. Los posibles efectos más suaves de algunos inductores leves en algunas de estas rutas endógenas están comenzando a describirse en la literatura.
ConclusiónEl autor propone que la literatura neuropsicofarmacológica sobre las interacciones farmacológicas en la epilepsia1 y el trastorno bipolar2 está seriamente contaminada en la actualidad por falsos negativos. Esta revisión detallada de la literatura sobre las propiedades inductivas de los FAE aporta recomendaciones prácticas a los clínicos, mientras que la parteii revisa los mecanismos farmacológicos que explican la inducción, y el modo en que los estudios futuros pueden mejorar las deficiencias actuales de la literatura.
Esta partei reconoce que incluso una revisión detallada de la literatura no puede eliminar la limitación de esta, que muestra una predisposición hacia los hallazgos falsos negativos. Al revisar la literatura, el autor ha tratado de combinar los enfoques de la MBE y de los mecanismos farmacológicos para elaborar las mejores recomendaciones para los clínicos en este momento. Sin embargo, el autor reconoce que es probable que en un plazo de 5años este artículo de revisión esté obsoleto, y que los factores de corrección incluidos (tabla 4) deban ser ampliamente modificados.
Existe un consenso general en la literatura33,34 acerca de que 3 FAE —carbamazepina, fenobarbital y fenitoína— son inductores clínicamente relevantes (tabla 2); además, todos los barbitúricos, incluyendo la primidona, deberían ser incluidos como tales. La tabla 4 incluye los factores de corrección calculados por el autor, utilizando la literatura disponible para corregir los efectos inductivos de la carbamazepina, el fenobarbital y la fenitoína. El autor precisa reconocer que el uso de factores de corrección constituye una mera simplificación para orientación a los clínicos, ya que existe gran variabilidad dentro de la población, en relación a los efectos inductivos123. El autor comenzó a utilizar factores de corrección hace un decenio3, debido a la falta de información práctica en las revisiones publicadas. Algunos de los factores de corrección son tan elevados que no deberían prescribirse otros fármacos junto con inductores potentes. Entre los antipsicóticos de segunda generación que no deberían prescribirse junto con inductores potentes se encuentran la lurasidona y la quetiapina. El bupropión es el caso más claro de un antidepresivo de segunda generación que no debería prescribirse conjuntamente con inductores potentes. La fenitoína, aparte de ser un inductor potente para múltiples enzimas, puede causar inhibiciones clínicamente importantes del CYP2C9 y el CYP2C19 mediante la saturación de dichas enzimas cuando las concentraciones plasmáticas de fenitoína son >20μg/ml.
El autor reconoce que la clasificación del clobazam, la eslicarbazepina, el felbamato, la lamotrigina, la oxcarbazepina, la rufinamida, el topiramato, la vigabatrina y el AVP como inductores leves no se había producido nunca en la literatura, pero se justifica porque contienen elementos comunes (tabla 5) y, más importantemente, pueden ayudar a los prescriptores a entender mejor este tema tan ignorado. Todos ellos tienden a tener efectos inductivos, a menudo combinados con propiedades inhibitorias (la vigabatrina constituye la excepción; tabla 6). Parecen requerir meses en alcanzar los efectos máximos, o en desaparecer, y definitivamente requerir más tiempo que los inductores potentes (tabla 5). Cinco inductores leves —el clobazam, la eslicarbazepina, el felbamato y la oxcarbazepina—, en dosis elevadas (≥1.200mg/día), y el topiramato en dosis elevadas (≥400mg/día), son inductores del CYP3A4 y pueden tener efectos clínicamente relevantes sobre los fármacos altamente dependientes del CYP3A4 para su metabolismo, incluyendo los anticonceptivos orales, los antipsicóticos tales como la lurasidona y la quetiapina, y las benzodiazepinas tales como el triazolam, el midazolam y el alprazolam. Las propiedades inductivas del AVP sobre el metabolismo de la clozapina y la olanzapina requieren más estudio, aunque el autor ha publicado información que establece claramente que puede ser clínicamente relevante en algunos pacientes.
Definitivamente, los inductores potentes, y posiblemente, los inductores leves tienen efectos relevantes sobre el metabolismo endógeno de: a)las hormonas sexuales; b)la vitaminaD; c)las hormonas tiroideas; y d)el metabolismo lipídico.
Conflicto de interesesNinguna organización comercial ha participado en la escritura de este documento o en su publicación. El autor reporta la ausencia de relación financiera, con intereses comerciales, durante los últimos 36 meses.
ReconocimientosEl autor reconoce la labor de Lorraine Maw, M.A., y Margaret T. Boden, R.N., M.L.T., del Centro de Investigación en Salud Mental en el Eastern State Hospital, Lexington, KY, Estados Unidos, por su colaboración para la edición de este artículo.