




Recently, we reported on a new MDD-like mouse model based on a regionally selective knockdown of astroglial glutamate transporters, GLAST/GLT-1, in infralimbic cortex (IL) which evokes widespread changes in mouse brain associated with the typical alterations found in MDD patients. To further characterize this new MDD-like mouse model, here we examine some transcriptional elements of glutamatergic/GABAergic neurotransmission and neuroplasticity in forebrain regions in the GLT-1 knockdown mice. Furthermore, we assess the acute ketamine effects on these transcriptional processes.
Material and methodsWe used a small interfering RNA (siRNA) pool targeting GLT-1 mRNA to disrupt the GLT-1 transcription in mouse IL. Histological assays were performed to examine postsynaptic density protein-95 (PSD95), neuritin (NRN), glutamine acid descarboxilase-65 (GAD65), and GLT-1 mRNA expression in IL and hippocampus.
ResultsKnockdown of GLT-1 in mouse IL leads to decreased expression of PSD95 and NRN neuroplasticity mRNAs in IL and hippocampus, which was reversed by an acute dose of ketamine antidepressant. Likewise, a single dose of ketamine also increased the mRNA levels of GAD65 and GLT-1 in IL of GLT-1 knockdown mice, reaching the basal values of control mice.
ConclusionsThe glutamatergic neuronal hyperactivity and deficits in the GABA system resulting from siRNA-induced astroglial glutamate transporter knockdown in IL can compromise the integrity/plasticity of neurocircuits affected in MDD. Suitable depressive-like animal models to address the neurobiological changes in MDD are an unmet need and the development of the GLAST/GLT-1 knockdown mouse model may represent a better option to understand the rapid-acting antidepressant effects of ketamine.
Recientemente, informamos sobre un nuevo modelo de ratón de depresión basado en una reducción parcial de la transcripción de los transportadores de glutamato, GLAST/GLT-1, en los astrocitos de la corteza infralímbica (IL), que conduce a cambios generalizados de la función cerebral del ratón, que refleja alteraciones típicas encontradas en pacientes con depresión. Para caracterizar más detalladamente este nuevo modelo de ratón de depresión, aquí examinamos algunos elementos transcripcionales relacionados con la neurotransmisión glutamatérgica/GABAérgica y la neuroplasticidad en regiones corticales y subcorticales de los ratones con niveles reducidos de GLT-1. Además, evaluamos los efectos agudos de la ketamina, antidepresivo de acción rápida, en estos procesos transcripcionales.
Material y métodosUtilizamos ARN de interferencia (siRNA) dirigido al mRNA de GLT-1 para interrumpir su transcripción en la IL de ratón. Se realizaron ensayos histológicos para examinar la expresión de los mRNA de las siguientes proteínas: proteína de densidad postsináptica-95 (PSD95), neuritina (NRN), descarboxilasa de ácido glutámico-65 (GAD65) y GLT-1 en la IL e hipocampo.
ResultadosLa reducción de la expresión de GLT-1 en IL de ratón conduce a una disminución de la expresión de los mRNA de neuroplasticidad PSD95 y NRN en la IL y el hipocampo, efecto que se revirtió con una única administración del antidepresivo ketamina. Asimismo, una sola dosis de ketamina también aumentó los niveles de los mRNA de GAD65 y GLT-1 en la IL de ratones silenciados para GLT-1, que alcanzaron los valores basales de los ratones control.
ConclusionesLa hiperactividad neuronal glutamatérgica y los déficits en el sistema GABAérgico resultantes de la reducción de los niveles del transportador de glutamato astroglial inducida por siRNA en IL pueden comprometer la integridad/plasticidad de aquellos neurocircuitos afectados en la depresión. Sin embargo, no disponemos de modelos animales de depresión adecuados para abordar los cambios neurobiológicos que se suceden en esta enfermedad y su tratamiento. El desarrollo del modelo de ratón con reducción parcial de GLAST/GLT-1 puede representar una mejor opción para comprender los efectos antidepresivos de acción rápida de la ketamina.
Major depressive disorder (MDD) is a heterogeneous illness that causes profound disability worldwide, affecting ∼7–12% of men and ∼20–25% of women.1 This situation is attributable to the high prevalence of MDD and to the slow clinical action and limited efficacy of monoamine-based antidepressants. Symptoms of MDD are associated with structural and synaptic plasticity changes, as well as neurochemical deficits in corticolimbic brain regions.2,3 Indeed, neuroimaging studies reported abnormalities of energy metabolism in ventral areas of anterior cingulate (vACC) in MDD patients.4,5 Although genetic/epigenetic factors confer some heritable risk of developing MDD, it is evident that exposure to repeated stressors may increase the vulnerability or even cause MDD in humans.6
Interestingly, astrocytes are emerging as key players in synaptic function, controlling extracellular levels of ions and neurotransmitters, responding to them, and releasing gliotransmittters that regulate synaptic transmission, plasticity, and animal behavior.7,8 The astroglial glutamate transporters, GLAST and GLT-1 (EAAT1 and EAAT2, respectively, in humans), take up most synaptic glutamate from central excitatory synapses, thereby exerting a tight direct control of neuronal excitability.9 Astrocytes have been proposed as an early contributor to the underlying pathogenesis of MDD,10 and alterations in astrocyte number/function have been reported in MDD patients and suicide victims.11,12 Hence, the functional hyperactivity reported in the vACC of MDD patients may result from a disturbed astrocyte-based glutamate homeostasis. In an attempt to mimic glial dysfunction in MDD, we developed a MDD-like mouse model based on small interfering RNA (siRNA)-induced knockdown of GLT-1/GLAST expression in infralimbic cortex (IL, rodent equivalent of vACC).13 Intra-IL siRNA targeting GLT-1/GLAST infusion evoked a moderate, long-lasting (≥7 days) decrease of GLAST/GLT-1 expression and number of astrocytes - reductions of ∼20–28% comparable to 24% described in MDD patient brains –12 resulting in enhanced excitatory neuronal activity in IL.14 Simultaneously, mice showed a robust depressive-like phenotype associated to a reduction of serotonin (5-HT) release and brain derived neurotrophic factor (BDNF) expression in the medial prefrontal cortex and hippocampus.13,14 The behavioral phenotype was reversed by citalopram and ketamine antidepressants.13
To further characterize this new MDD-like mouse model, here we extend the above observations to examine the transcriptional elements of glutamatergic/GABAergic neurotransmission and neuroplasticity in forebrain regions in GLT-1 knockdown mice. We pay special attention to postsynaptic density protein-95 (PSD95) and neuritin (NRN), both downstream synaptic plasticity targets regulated by BDNF/TrkB signaling pathway,3 which is impaired in the GLT-1 knockdown mouse model. We also assess the acute ketamine effects on these transcriptional processes.
Materials and methodsAnimalsMale C57BL/6J mice (14 weeks; n=24 for the whole study, Charles River, Lyon, France) were housed under controlled conditions (22±1°C; 12h light/dark cycle) with food and water available ad-libitum. Animal procedures were conducted in accordance with standard ethical guidelines (EU directive 2010/63 of 22 September 2010) and approved by the local ethical committee (University of Barcelona).
Treatments and siRNA synthesisSynthesis and purification of siRNA sequences targeting GLT-1 (GLT-1-siRNA: GenBank accession NM_001077514) were performed by nLife Therapeutics S.L. (Granada, Spain). Mice were anesthetized with pentobarbital (40mg/kg, i.p.) and were placed in the stereotaxic frame. Mice received unilaterally 1μl of artificial cerebrospinal fluid (aCSF: 125mM NaCl, 2.5mM KCl, 1.26mM CaCl2 and 1.18mM MgCl2 with 5% glucose) or GLT-1-siRNA into IL (coordinates anterior–posterior: +2, medial–lateral: −0.2, dorsal–ventral: −3.3 in mm). The siRNA infusion was performed using a glass capillary attached to a 10μl Hamilton syringe connected to a precision minipump (KDS 310 plus syringe pump) at a rate of 0.3μl/min. GLT-1-siRNA pool was prepared in RNAse-free aCSF and infused at a dose of 60μg/μl (20μg of each sequence).13,14 To examine the ketamine effects on mRNA expression profile in forebrain of GLT-1 knockdown mice, a single dose of saline (0.9%) or ketamine (10mg/kg, i.p.) was administered 24h after GLT-1-siRNA infusion. Mice were euthanized 2h or 24h later (Fig. 1a).
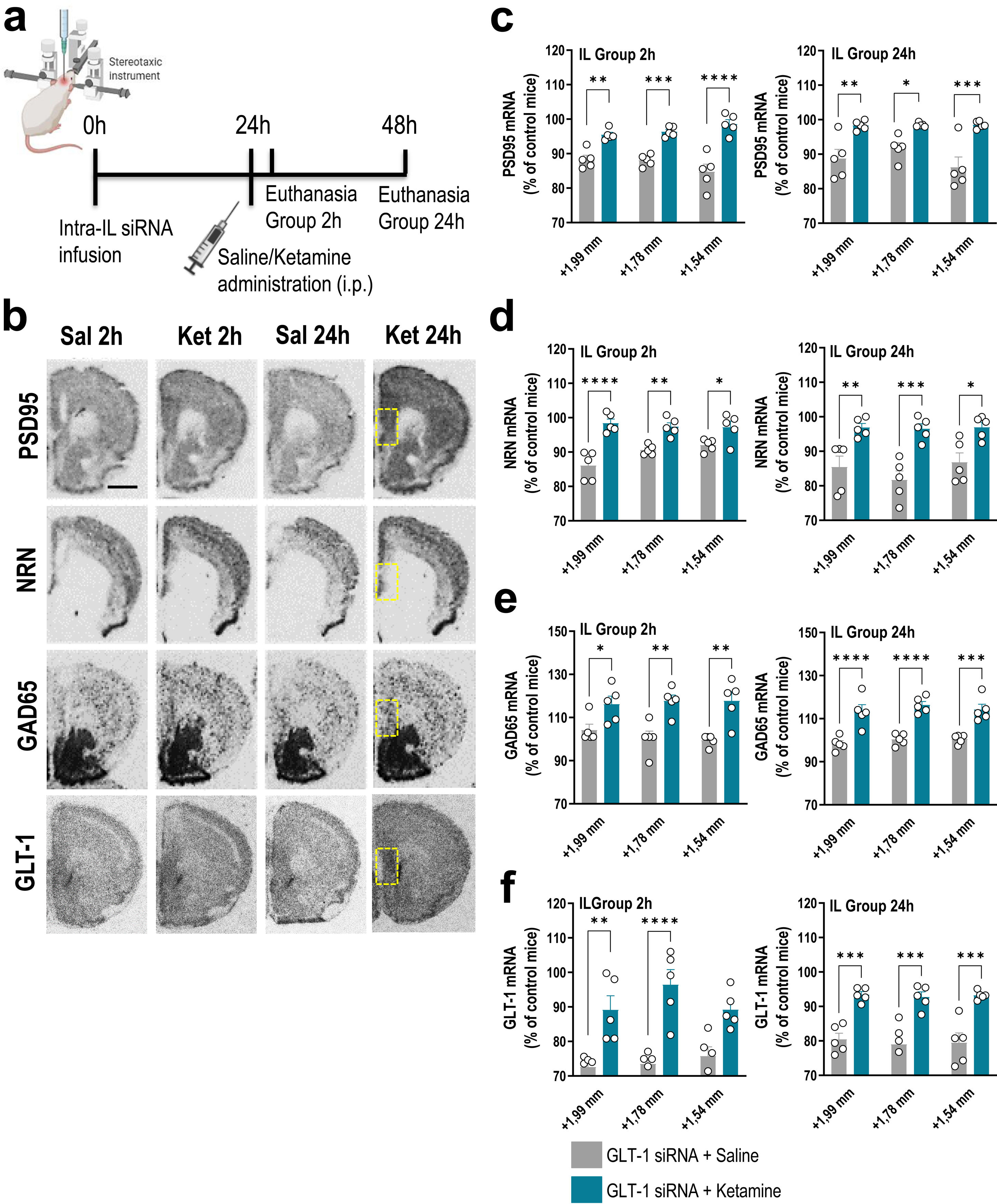
 Treatment schedule. Mice were infused with GLT-1-siRNA (60μg/μl) or vehicle into IL and 24h later, they received saline or ketamine (10mg/kg, i.p.). Mice were euthanized at 2h or 24h after saline/ketamine administration. (b) Representative coronal brain sections showing PSD95 (postsynaptic density protein-95), NRN (neuritin), GAD65 (glutamic acid descarboxilase-65) and GLT-1 mRNA expression in the medial prefrontal cortex of mice assessed by in situ hybridization. The yellow frames show the IL section in which the different mRNAs were analyzed. Scale bar: 1mm. (c–f) Densitometric analysis of PSD95 (c), NRN (d), GAD65 (e), and GLT-1 (f) mRNA levels in IL of GLT-1 knockdown mouse model treated with saline or ketamine at 2h and 24h post-administration. A single dose of ketamine rapidly increased the expression of PSD95, NRN, GAD65, and GLT-1 mRNAs in the different anteroposterior coordinates of the IL (from +1.99 to −+1.54mm). This effect was sustainable 24h later. Data are presented as the mean±SEM (n=5 mice per group). *p<0.05, **p<0.01, ***p<0.001 and ****p<0.0001 compared to GLT1-siRNA-infused mice treated with saline.")
A single dose of ketamine causes rapid transcriptional changes in IL of GLT-1 knockdown mouse model. (a) Treatment schedule. Mice were infused with GLT-1-siRNA (60μg/μl) or vehicle into IL and 24h later, they received saline or ketamine (10mg/kg, i.p.). Mice were euthanized at 2h or 24h after saline/ketamine administration. (b) Representative coronal brain sections showing PSD95 (postsynaptic density protein-95), NRN (neuritin), GAD65 (glutamic acid descarboxilase-65) and GLT-1 mRNA expression in the medial prefrontal cortex of mice assessed by in situ hybridization. The yellow frames show the IL section in which the different mRNAs were analyzed. Scale bar: 1mm. (c–f) Densitometric analysis of PSD95 (c), NRN (d), GAD65 (e), and GLT-1 (f) mRNA levels in IL of GLT-1 knockdown mouse model treated with saline or ketamine at 2h and 24h post-administration. A single dose of ketamine rapidly increased the expression of PSD95, NRN, GAD65, and GLT-1 mRNAs in the different anteroposterior coordinates of the IL (from +1.99 to −+1.54mm). This effect was sustainable 24h later. Data are presented as the mean±SEM (n=5 mice per group). *p<0.05, **p<0.01, ***p<0.001 and ****p<0.0001 compared to GLT1-siRNA-infused mice treated with saline.
Mice were killed by pentobarbital overdose and brains were rapidly removed, frozen on dry ice and stored at −80°C. Coronal tissue sections (14μm-thick) were cut using a microtome-cryostat (HM500-OM, Microm, Germany), thaw-mounted onto 3-aminopropyltriethoxysilane (Sigma–Aldrich)-coated slides and kept at −20°C until use, as previoulsy reported.13 Antisense oligoprobes were complementary to bases: PSD95/76-120 (GenBank accession NM_007864), NRN/408-448 (GenBank accession NM_153529), glutamic acid decarboxylase-65 GAD65/776-820 (GenBank accession NM_000818.2), and GLT-1/666-716 (GenBank accession NM_011393), and synthethized by Ibian Technologies (Zaragoza, Spain). Oligonucleotides were individually labeled (2pmol) at the 3′-end with [33P]-dATP (>2500Ci/mmol; DuPont-NEN, Boston, MA) using terminal deoxynucleotidyltransferase (TdT, Calbiochem, La Jolla, CA).13 Hybridized sections were exposed to Biomax-MR film (Kodak, Sigma–Aldrich, Madrid, Spain) for 2 days. For specificity control, adjacent sections were incubated with an excess (50×) of unlabeled probes.
Films were analyzed and relative optical densities (ROD) were obtained using a computer-assisted image analyzer for image acquisition (Micro-manager 1.4, San Francisco, CA). The slide background and non-specific densities were subtracted. ROD was evaluated in 2–3 duplicate adjacent sections from each mouse and averaged to obtain individual values along the anteroposterior axis (three consecutive and equidistant measures between anterior–posterior coordinates +1.98 to +1.54mm for IL) using the Fiji/ImageJ software. Figures were prepared for publication, in which contrast and brightness of images were the only variables adjusted digitally.
Statistical analysisResults are given as mean±SEM (n=4–5 mice/group). Statistical analyses were performed by two-way ANOVA followed by Tukey's post-hoc test as appropriate (GraphPad Prism 9.01; San Diego, CA). Before the analysis, all data was checked for normality (Anderson–Darling; D’Agostino–Pearson omnibus; Shapiro–Wilk; Kolmogorov–Smirnov) and homogeneity of variances (Bartlett's test and Brown–Forsythe test). Outlier values were identified by the Grubbs’ test (i.e., Extreme Student zed Deviate, ESD, method) using GraphPad Prism software and excluded from the analysis when applicable. Statistical significance has been set at the 95% confidence level.
ResultsPreviously, we showed that a single dose of non-competitive NMDA receptor antagonist ketamine (10mg/kg, i.p.) reversed MDD-like behavioral phenotype in GLT-1 knockdown mice.13 In order to examine whether ketamine may induce rapid changes on transcriptional processes related to synaptic plasticity, we assessed PSD95 and NRN mRNA expression in IL and hippocampus of GLT-1 knockdown mice, as previously reported.3
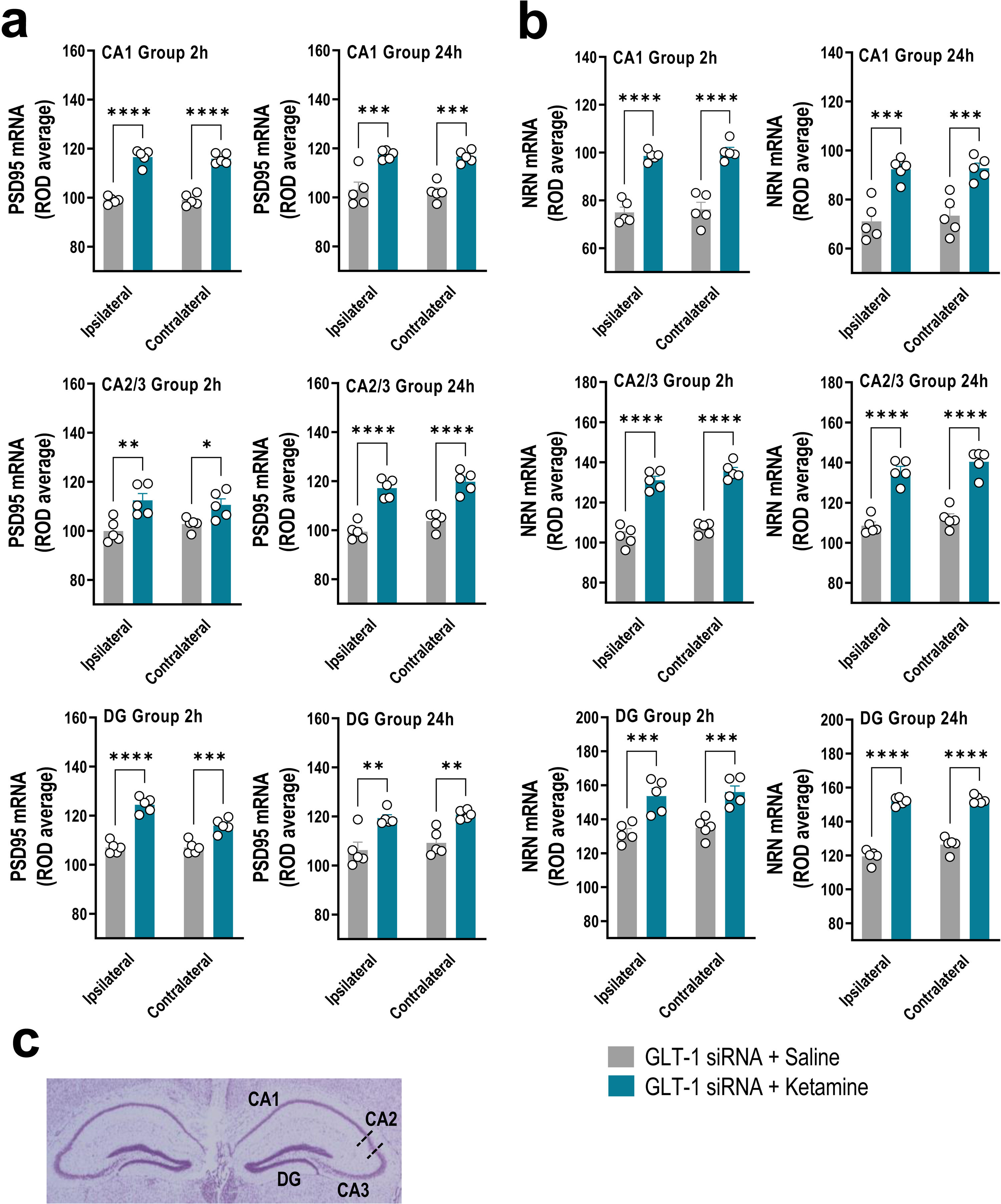
GLT-1 knockdown mice were treated with saline or ketamine (10mg/kg, i.p.) and euthanized at 2h or 24h later (Fig. 1a). This dose of ketamine rapidly increased the PSD95 and NRN mRNA levels along the anterior–posterior axis of IL at 2h and 24h later compared to GLT-1-siRNA-infused mice treated with saline, as assessed by ISH (Fig. 1b–d). Two-way ANOVA revealed an effect of group for PSD95 [F(1,8)=71.84, p<0.0001 and F(1,8)=42.58, p<0.0001], and for NRN [F(1,8)=95.68, p<0.0001 and F(1,8)=23.83, p<0.001] at 2 and 24h, respectively. Similarly, ketamine administration also increased PSD95 and NRN expression in the different regions of the hippocampus, e.g. CA1, CA2/3, and dentate gyrus (DG), compared to GLT-1-siRNA mice treated with saline (Fig. 2a–c).
 Bar graphics showing sustainable increases of PSD95 (a) and NRN (b) mRNA levels in both ipsilateral and contralateral sides of the hippocampal formation including CA1, CA2–CA3, and dentate gyrus (DG) in GLT-1 knockdown mouse model treated with ketamine compared to those treated with saline. (c) Representative cresyl violet-stained section showing different mouse hippocampal regions: CA1, CA2, CA3, and DG in which densitometry analyses were performed. Data are presented as the mean±SEM (n=5 mice per group). *p<0.05, **p<0.01, ***p<0.001 and ****p<0.0001 compared to GLT1-siRNA-infused mice treated with saline.")
Acute ketamine administration increased the neuroplasticity gene expression in the hippocampus of GLT-1 knockdown mouse model. (a, b) Bar graphics showing sustainable increases of PSD95 (a) and NRN (b) mRNA levels in both ipsilateral and contralateral sides of the hippocampal formation including CA1, CA2–CA3, and dentate gyrus (DG) in GLT-1 knockdown mouse model treated with ketamine compared to those treated with saline. (c) Representative cresyl violet-stained section showing different mouse hippocampal regions: CA1, CA2, CA3, and DG in which densitometry analyses were performed. Data are presented as the mean±SEM (n=5 mice per group). *p<0.05, **p<0.01, ***p<0.001 and ****p<0.0001 compared to GLT1-siRNA-infused mice treated with saline.
Furthermore, acute ketamine also enhanced gene expression related to components of glutamatergic/GABAergic neurotransmission in GLT-1 knockdown mice. Increased GAD65 mRNA expression was detected in IL of GLT-1-siRNA-infused mice treated with ketamine vs. GLT-1-siRNA-infused mice treated with saline (Fig. 1b, e). This effect was sustainable until 24h after ketamine administration. Two-way ANOVA revealed an effect of group for GAD65 mRNA at 2h [F(1,8)=17.89, p<0.01] and 24h [F(1,8)=87.64, p<0.0001]. In parallel, ketamine administration recovered the GLT-1 mRNA levels in GLT-1 knockdown mice resulting in an increased IL GLT-1 expression compared to GLT-1-siRNA-infused mice received saline (Fig. 1b, f). Two-way ANOVA revealed an effect of group for GLT-1 mRNA at 2h [F(1,8)=41.72, p<0.001] and 24h [F(1,8)=53.12, p<0.001]. Overall, these results argue that the efficacy of ketamine as a fast-acting antidepressant may occur through an improvement of glutamate/GABAergic balance in IL (vACC in humans), and the subsequent enhancement in synaptic plasticity.
DiscussionRecently, we reported on a new MDD-like mouse model based on a regionally selective knockdown of astroglial glutamate transporters, GLAST/GLT-1, in IL which evokes widespread changes in mouse brain associated with the typical alterations found in MDD patients.13,14 This was achieved by selective siRNA sequences targeting GLAST or GLT1 mRNAs and, although siRNAs may have off-target effects,15 we previously reported that GLT-1-siRNA does not affect GLAST expression and vice-verse or that of neuronal glutamate transporters (EAAC1 and vGLUT1), showing an excellent target specificity. Moreover, the same amount of nonsense siRNA infused into IL did not change GLT-1 expression.13 The regional and target selectivity of the present procedure is, in part, due to the use of low siRNA concentrations, consistent with previous observations, which allowed the knockdown of genes expressed by 5-HT neurons in mouse dorsal raphe nucleus, without affecting the neighboring median raphe nucleus.15 Overall, our results demonstrated a selective and sustained (at least for 7 days) reduction of GLT-1 mRNA expression (20–30%) in IL of mice that mimics a comparable decrease of human EAAT2 (mouse GLT-1) mRNA levels (30–50%) in vACC and dorsolateral prefrontal cortex of MDD patients.16
In the present study, we showed that this new MDD-like mouse model also leads to changes in neuroplasticity markers, such as reduced PSD95 and NRN mRNA expression in IL and hippocampus, perhaps associated with widespread fall in brain BDNF expression, as previously found.13 Hence, some mechanisms that could be underlying the rescue of the depressive-like phenotype triggered by ketamine in the GLT-1 knockdown mice13 would include activation of the PSD95 and NRN expression, as reported herein. NRN is a member of the neurotrophic factor family enriched at the synapse, which promotes neurite outgrowth and branching, and BDNF/TrkB signaling regulates its expression. Similarly, PSD95 induction is consistent with increased synapse formation and function, involving complex BDNF actions that promote the formation of new synapses. Both NRN and PSD95 were found to be reduced in postmortem brain samples from MDD patients and in stress-induced depressive-like rodent models, while antidepressant treatments increased them.2,3 Although the mechanisms underlying the rapid antidepressant effects of ketamine are unclear, a main hypothesis is that ketamine increases the synaptic glutamate neurotransmission, inducing BDNF release and mTOR activation, which in turn increases the synthesis of synaptic proteins. Therefore, ketamine-induced activation of these molecular pathways would lead to enhancement expression of NRN and PSD-95 in the forebrain of GLT-1 knockdown mice, suggesting that both components are critical downstream mediators of antidepressant ketamine effects.
Early hypothesis on MDD focused on a reduced activity of brainstem monoaminergic systems (mainly 5-HT and norepinephrine, NE). One commonality of these ascending monoamine systems is that they are densely innervated by descending afferents from the PFC, which tightly control their activity.17 Therefore, given the top-down control exerted by the PFC on 5-HT/NE activity and the widespread brain innervation by these monoamine systems, a focal alteration of neuronal activity in a restricted PFC area (e.g., unilateral IL) may translate into a global change in brain activity resulting from monoamine hypofunction, and leading to the multiple and varied depressive symptomatology. Supporting this hypothesis, at least regarding 5-HT, the selective reduction of GLT-1 expression in IL evoked a very marked reduction of 5-HT release in the DR, and decreased BDNF expression, not only in IL but also in subcortical areas such as the ipsilateral and contralateral hippocampal formation.13 According to these data, bilateral alterations in PSD95 and NRN expression were also detected in GLT-1 knockdown mice, which may be driven by changes in BDNF expression.
Recently, reduced hippocampal GLT-1 levels were reported in stress-based animal models, and these were restored by the antidepressants fluoxetine and ketamine.18 Based on these data, a single dose of ketamine reversed the depressive-like behavioral phenotype,13 while GLT-1 mRNA increased in GLT-1 knockdown mice reaching control values, suggesting that GLT-1 as a potential therapeutic target for MDD. Likewise, several studies showed dysregulation of GABA neurotransmission and decreased GABA concentrations in MDD patients and stress- or genetic-based animal models of depression. In addition, the GABA system has become a therapeutic target for fast-acting non-monoaminergic pharmacological approaches to treat MDD, including GABAA and GABAB receptor modulators (allosteric modulators, neurosteroids, agonists, and antagonists), ketamine, and neuropeptides targeting GABAergic interneurons.19 Consistent with these observations, a single dose of ketamine was sufficient to enhance the GAD65 transcription, one of the GABA synthesizing enzyme, in GLT-1 knockdown mice. The increased expression of GAD65 after ketamine, probably reflects the deficiency of the GABA system reported in MDD patients and depressive-like animal models, including GLT-1 knockdown model.
In conclusion, this study allowed a more in-depth characterization of the new MDD-like mouse model based on altered glutamatergic homeostasis in IL, shedding some light on the putative mechanisms related to glutamate/GABA balance and neuroplasticity that could underlie the fast-acting antidepressant effects of ketamine. Suitable MDD-like animal models to address neurobiological changes in MDD are an unmet need and the development of the GLAST/GLT-1 knockdown mouse model may represent a better option.
FundingThis study was supported by grants SAF2016-75797-R, PID2019-105136RB-100, Ministry of Economy and Competitiveness (MINECO) and European Regional Development Fund (ERDF), UE; and CB/07/09/0034Center for Networked Biomedical Research on Mental Health (CIBERSAM).
Conflict of interestThe authors declare that they have no conflict of interest.
We thank to Spanish Ministry of Education, Culture, and Sport for their financial support via a scholarship awarded to M.N.F