



El síndrome de Vogt-Koyanagi-Harada (VKH), también conocido como síndrome uveomeníngeo, es una panuveítis granulomatosa bilateral y difusa que cursa con desprendimiento de retina seroso y que puede acompañarse de afectación del sistema nervioso central, alteraciones dermatológicas y auditivas.
Su nombre deriva de los autores que la describieron por primera vez. Afecta a adultos de ambos géneros, entre los 20 y 50 años de edad, y presenta una prevalencia aumentada en razas negras. Este síndrome inflamatorio probablemente sea el resultado de un mecanismo autoinmune, influenciado por factores genéticos.
La evolución de la enfermedad se divide en 4 estadios clínicos: prodrómico, uveítico agudo, de convalecencia y crónico recurrente. El diagnóstico es fundamentalmente clínico, mediante los criterios establecidos por la Sociedad Americana de Uveítis (AUS) publicados en el año 2001. Es necesario realizar diagnóstico diferencial con la oftalmía simpática, el linfoma primario de células B, la escleritis posterior y el síndrome de efusión uveal. El tratamiento precoz y mantenido es la base de una buena evolución.
The Vogt-Koyanagi-Harada (VKH) disease, formerly known as uveomeningitic syndrome, is a bilateral diffuse granulomatous panuveitis associated with exudative retinal detachment, which can be accompanied by central nervous system involvement, dermatological and auditory alterations.
Its name derives from the authors who first described the disease. It affects adults of both genders, between 20 and 50 years old, with darkly pigmented races prevalence. This inflammatory syndrome is probably the result of an autoimmune mechanism, influenced by genetic factors.
The evolution of the disease is divided into four clinical stages: prodromal, acute uveitic, convalescent and chronically recurrent. The diagnosis is mainly clinical, using the criteria established by the American Society of Uveitis (AUS) posted in the year 2001. Differential diagnosis must be done with sympathetic ophthalmopathy, primary B cell lymphoma, posterior scleritis, and uveal effusion syndrome. An early and maintained treatment is the basis of a favorable outcome.
Desde la antigüedad existen en la literatura numerosas referencias de esta patología. La primera data del siglo x AC y corresponde al persa Ali Ibn Isa, quien describió el blanqueamiento de las pestañas, cejas y cabello asociado a inflamación ocular.
Fue Vogt quien en 1906 refirió un caso de uveítis anterior asociado con poliosis y disacusia1. En 1926, Harada describió la presencia de uveítis posterior con desprendimiento de retina exudativo, asociado a signos meníngeos y pleocitosis del líquido cefalorraquídeo1. En 1929, Koyanagi describió a pacientes con iridociclitis bilateral asociada a alopecia y vitíligo, sordera y tinnitus.
No fue hasta la década de los cuarenta cuando Babel reconoció el síndrome de Vogt-Koyanagi-Harada (VKH) o síndrome uveomeníngeo como una única entidad.
EpidemiologíaAfecta a adultos de ambos géneros, con algo más de predilección por el género femenino, entre los 20 y 50 años de edad. La incidencia y prevalencia no son bien conocidas. Presenta una predilección por razas pigmentadas (orientales, negros e hispanos) y muy baja por razas blancas. Según diferentes series, se ha visto que la prevalencia oscila entre el 4 y el 8% del total de uveítis endógenas.
EtiopatogeniaAunque la etiología es desconocida, se cree que podría tener un origen autoinmune. Existe mayor asociación a grupos étnicos de piel oscura, aunque la pigmentación no lo explica todo, ya que esta patología es infrecuente en africanos subsaharianos.
Se ha sugerido que existiría una reacción inmune de tipo directo contra antígenos de células que contengan melanina, como consecuencia de una lesión cutánea o de una infección, en personas genéticamente susceptibles.
La respuesta vendría mediada por linfocitos T y dirigida contra antígenos melanocíticos del ojo (concretamente de la retina e iris), la piel, el sistema nervioso y el oído interno. Entre las posibles dianas antigénicas se han descrito la tirosinasa y la proteína S-1001. En estudios con microscopio electrónico se ha visto contacto íntimo entre melanocitos y linfocitos de la membrana basal coroidea2.
En el ojo se produce una reacción inflamatoria uveal granulomatosa con presencia de linfocitos y células epitelioides. En fases agudas se ha visto una preponderancia de linfocitos CD4+, mientras que en fase de convalecencia lo son los CD8+3. En fases crónicas se observa la desaparición de los melanocitos coroideos y alteraciones del epitelio pigmentario de la retina.
Existe una asociación a diferentes haplotipos del HLA, y el más frecuente es el HLA-DR4. Recientemente se ha definido un nuevo autoantígeno del síndrome, el KU-MEL-1, formando parte HLADRB1, que podría ser utilizado para el diagnóstico de la enfermedad4.
Manifestaciones clínicasLa enfermedad se presenta en 4 fases distintas:
Fase prodrómicaSuele durar desde días a semanas, generalmente una semana. Se presenta en forma de clínica seudogripal con fiebre, cefalea, náuseas y dolor orbitario. Suele cursar con tinnitus y, menos frecuentemente, con manifestaciones neurológicas.
Fase uveítica agudaSe inicia unos días después de la fase prodrómica y puede durar entre 2 y 3 meses. Se caracteriza por una uveítis bilateral, aunque en algunos casos puede afectarse primero un ojo y posteriormente bilateralizarse, con la aparición súbita de visión borrosa, fotofobia y dolor ocular. Puede acompañarse de disacusia y meningismo.
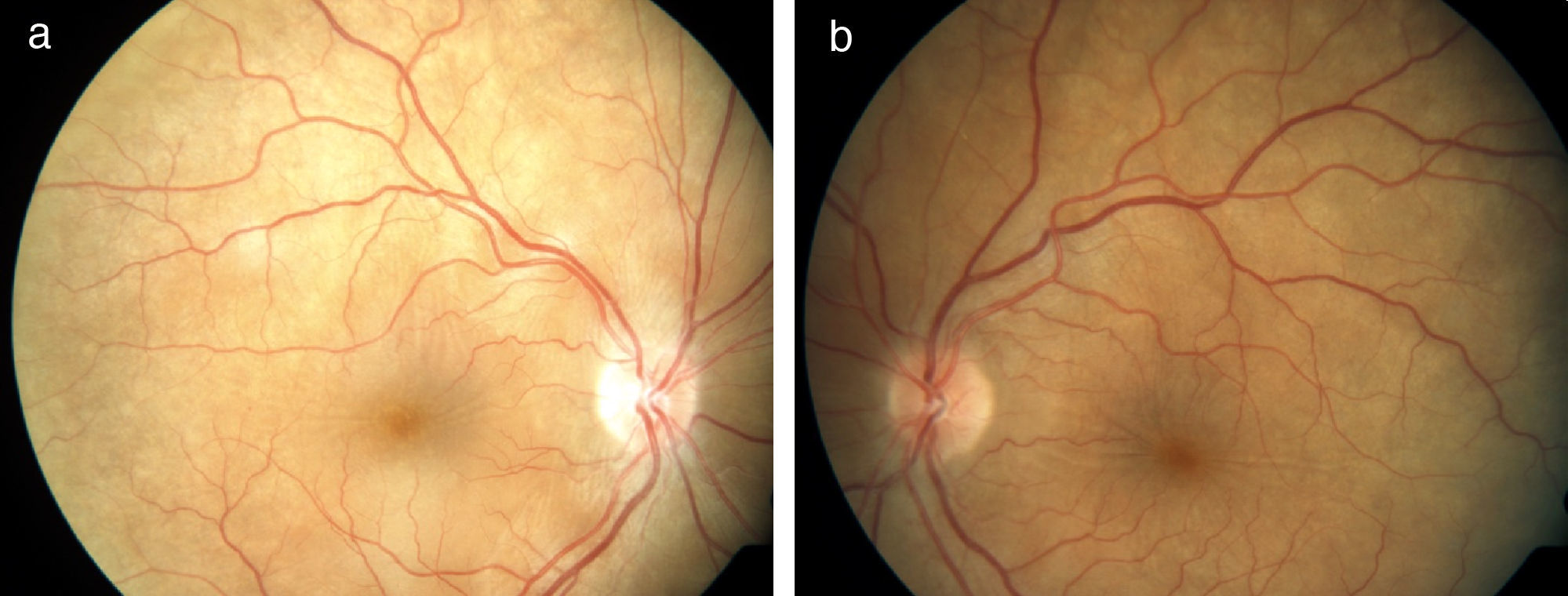
La afectación predomina en el polo posterior, pudiéndose observar edema de papila con desprendimiento seroso de retina (fig. 1). La inflamación puede extenderse al polo anterior presentando precipitados queratínicos corneales, pequeños nódulos en el borde pupilar (llamados nódulos de Koeppe) e incremento de la presión intraocular (PIO). En algunos casos puede presentarse de forma sinequiante, aumentando el riesgo de incremento de la PIO. Todos estos signos son manifestaciones de una reacción granulomatosa en la úvea ocular (panuveítis). Si la inflamación ocular es acusada, se hace muy manifiesta la hipoacusia central (75%), especialmente a frecuencias altas.
. Se observan abundantes pliegues maculares que son compatibles con desprendimiento de retina exudativo en ambos ojos. La papila es de aspecto normal.")
En esta fase se ponen de manifiesto algunos trastornos endocrinológicos dados por inflamación hipotalámica, tales como amenorrea, disfunción hipofisaria, diabetes insípida, hiperglucemia e hipocolesterolemia5.
La sintomatología puede confundirse con hipertensión intracraneana, dado que se acompaña de cefalea y vómitos. Sin embargo, la presión intracraneana en general es normal y solo en un pequeño porcentaje se encuentra sutilmente elevada.
Fase crónica o convalecenciaAparece a los 3 meses de la fase aguda. En ella son más frecuentes los síntomas cutáneos, aunque estos pueden aparecer antes, durante o después del compromiso ocular. Se han descrito canicie, poliosis, alopecia y vitíligo, que en algunas ocasiones se presenta siguiendo una distribución neural, acompañándose de parestesias y disestesias. Afecta frecuentemente a la cara, el cuello, el tronco y los párpados.
En cuanto a la afectación ocular, se observa una despigmentación uveal progresiva con coloración rojo anaranjada conocida como sunset glow fundus, más frecuente en asiáticos. Suele aparecer unos 3 meses después de la afectación uveal. También se han descrito alteraciones del epitelio pigmentario de la retina (redistribución de pigmento que forman imágenes similares a nevus), áreas atróficas denominadas atrofia circumpapilar coriorretiniana, e hiperpigmentarias difusas, que son las que se describen en las razas hispanoamericanas. En el limbo esclerocorneal se puede encontrar una despigmentación conocida como «signo de Sugiura» o vitíligo perilímbico, que se limita casi exclusivamente a la población japonesa.
Fase crónica recurrenteTiene lugar a los meses-años después de la fase aguda, interrumpiendo la fase de convalescencia. Esta fase no está presente en todos los pacientes. Se caracteriza por episodios de uveítis anterior granulomatosa frecuentemente resistente a tratamiento esteroideo. De forma infrecuente se asocia a uveítis posterior. Característicamente suelen aparecer nódulos de iris.
Es la fase de las complicaciones crónicas, como cataratas, glaucoma, edema macular, neovascularización coroidea y papilar y membrana neovascular subretiniana.
Manifestaciones extraoculares del síndrome de Vogt-Koyanagi-HaradaEl compromiso neurológico puede aparecer antes, durante o después de la afectación ocular y tener una localización y gravedad variables. Cursa con hiperestesia cutánea hasta en el 70% de los casos, y se evidencia cuando el paciente se peina. De forma más infrecuente se asocia a focalidades neurológicas como hemiparesia, ataxia cerebelosa, mielitis transversa, disartria, afasia o psicosis. Los pares craneales que más frecuentemente se afectan son el II y el VIII, pudiendo también comprometerse el III, el IV, el V, el VI y el VII. En el 80% de los casos cursa con meningitis linfocitaria, que puede durar hasta 8 semanas.
Las manifestaciones auditivas se presentan hasta en el 30% de los casos por compromiso del VIII par, en forma de disacusia central a frecuencias altas, acufenos, tinnitus y sordera neurosensorial que se caracteriza por ser rápida, progresiva y bilateral. Algunas veces la pérdida de la audición al inicio puede ser unilateral, asimétrica, fluctuante y coincidente con los episodios de brote.
El componente vestibular condiciona vértigo en el 50% de los casos, y de forma más infrecuente nistagmo horizontal, alteración del reflejo oculovestibular y de los movimientos oculares del seguimiento lento1,6.
Las manifestaciones dermatológicas suelen incluir alopecia en más del 70% de los pacientes, poliosis o vitíligo, más frecuentemente encontrados en la fase de convalecencia.
Se describen 3 tipos de síndrome de VKH:
- •
Tipo I: hay compromiso ocular sin compromiso auditivo ni cutáneo, que permite sospechar el VKH.
- •
Tipo II: hallazgos oculares, con al menos una manifestación auditiva o cutánea.
- •
Tipo III: el paciente presenta afectación oftalmológica, auditiva y cutánea.
Aproximadamente el 70% de los casos del tipo I y II tienen una duración de la enfermedad de menos de un año, mientras que dos tercios de los casos del tipo III presentan enfermedad activa por más de un año.
No hay relación entre la gravedad del compromiso visual y la de la enfermedad general, pero los pacientes con afectación tipo III tienen curiosamente mejor agudeza visual final que aquellos de tipo I y II5,7.
DiagnósticoEl diagnóstico se realiza por criterios clínicos establecidos por la Sociedad Americana de Uveítis (AUS), publicados en el año 2001 (tabla 1)8-10. El hallazgo ocular más característico es el desprendimiento de retina exudativo multifocal.
Criterios diagnósticos de Vogt-Koyanagi-Harada
| 1. Sin antecedentes de traumatismos perforantés o cirugías previas |
| 2. Sin evidencia de enfermedades oculares previas |
| 3. Afectación ocular bilateral (A o B, según el estadio de la enfermedad) |
| A. Manifestaciones tempranas |
| • Presencia de coroiditis difusa (áreas focales de líquido subretiniano o desprendimento de retina seroso bulloso) |
| • Si las lesiones no son tan evidentes, debe haber: áreas de retraso de relleno coroideo (AFG), áreas placoides de hiperfluorecencia y tinción de nervio óptico |
| • Engrosamiento difuso sin escleritis posterior (ecografía) |
| B. Manifestaciones tardías |
| • Antecedentes de las manifestaciones previas |
| • Despigmentación ocular: signo de sugiura o sunset glow fundus |
| • Otros, como cicatrices numulares coriorretinianas, uveítis anterior recurrente o crónica |
| 4. Antecedente o hallazgos neurológicos y/o auditivos |
| • Meníngismos |
| • Tinnitus y/o hipoacusia |
| • Pleocitosis en líquido cefalorraquídeo |
| 5. Hallazgos dermatológicos, posterior a la uveítis |
| • Alopecia, vitíligo, poliosis |
Los exámenes complementarios no serían necesarios pero nos aportan mayor certeza diagnóstica. La angiografía fluoresceínica en la fase aguda nos muestra múltiples spots hiperfluorescentes en el epitelio pigmentario. La ecografía ocular ayuda cuando hay medios opacos, mostrando un engrosamiento coroideo posterior o desprendimiento de retina seroso. La punción lumbar mostraría pleocitosis del líquido cefalorraquídeo con predominio de linfocitos, aunque si se muestran todos los signos y síntomas, esta no sería necesaria.
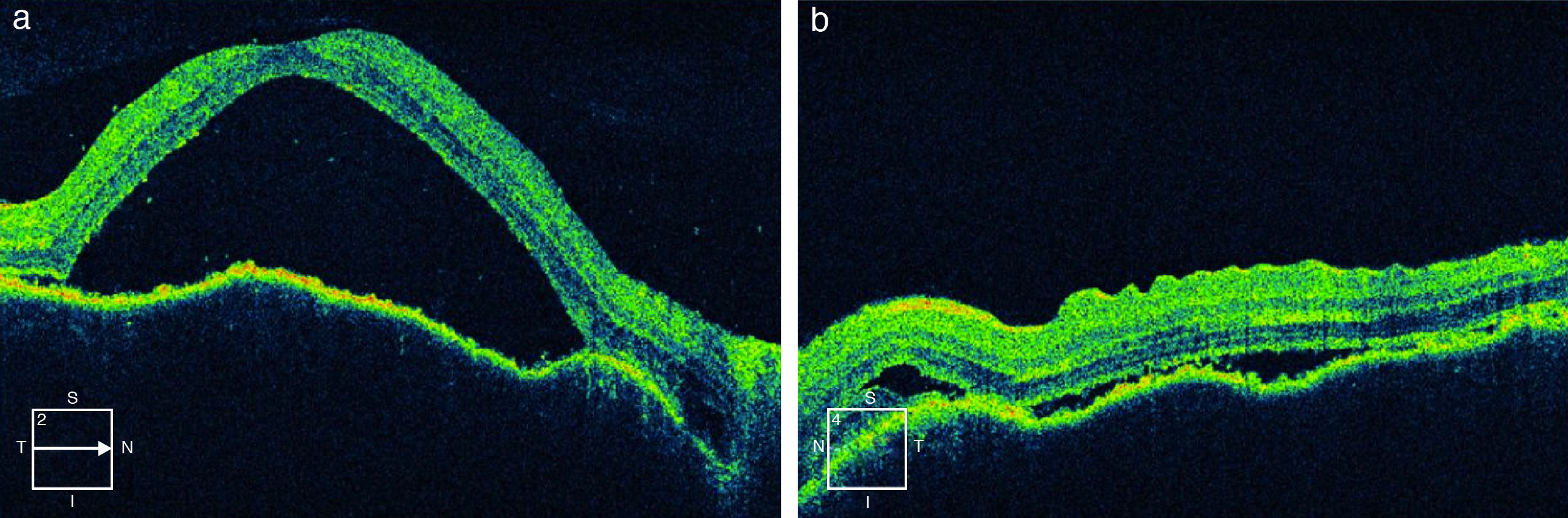
La RM permite diferenciar esta enfermedad de la escleritis posterior. La tomografía de coherencia óptica es útil para cuantificar la evolución del desprendimiento seroso, para el diagnóstico y seguimiento de complicaciones como el edema macular y las membranas neovasculares9,10 (fig. 2).
. Desprendimiento exudativo de retina. Se observa separación entre el epitelio pigmentario de la retina y la retina neurosensorial con fluido subretiniano en su interior.")
Tomografía de coherencia óptica en paciente con síndrome de Vogt-Koyanagi-Harada precoz (a: ojo derecho, b: ojo izquierdo). Desprendimiento exudativo de retina. Se observa separación entre el epitelio pigmentario de la retina y la retina neurosensorial con fluido subretiniano en su interior.
Se define como síndrome de VKH completo cuando el paciente cumple los criterios del 1 al 5, incompleto si se cumplen criterios del 1 al 3, más el cuarto o quinto criterio, y probable cuando solo están presentes los criterios oculares del 1 al 38-10.
Diagnóstico diferencialEl diagnóstico diferencial del síndrome VKH debe incluir otras entidades que cursen con uveítis posterior, panuveítis y con desprendimiento exudativo de retina, tales como la oftalmia simpática, síndromes de puntos blancos como la epiteliopatía placoide multifocal aguda o síndrome de múltiples puntos blancos evanescentes, el síndrome de efusión uveal, la coroidopatía geográfica, la escleritis posterior, la sarcoidosis, el linfoma primario intraocular de células B8.
Respecto a los síntomas cutáneos, hay que considerar posibilidades como el síndrome de Alezzandrini, la alopecia areata y el piebaldismo.
Además, deberíamos tener en cuenta en el diagnóstico diferencial la afectación neurosensorial del lupus eritematoso sistémico, la artritis reumatoide, el síndrome de Wegener, la colitis ulcerosa, la amiloidosis, la sarcoidosis y el síndrome de Cogan. No obstante, manifestaciones sistémicas como signos meníngeos, alteraciones auditivas y cambios pigmentarios son característicos de la enfermedad de VKH10.
PronósticoLa edad de comienzo, la duración de la fase crónica y la presencia de membranas neovasculares subretinianas son los principales factores pronósticos.
Es una enfermedad con relativo buen pronóstico y que mantiene visiones de 20/30 en el 50-60% de los pacientes. El pronóstico visual es muy variable, pero en general es bueno si el diagnóstico es temprano y se prescribe un tratamiento adecuado de forma mantenida en el tiempo. A pesar de ello, el 7% presenta resistencia al tratamiento, y en estos casos el pronóstico visual es infausto.
ComplicacionesEl 50% de los pacientes presentan complicaciones que incluyen, como ya mencionamos anteriormente: formación de cataratas en aproximadamente el 11-38% de los pacientes; glaucoma en el 40% de los pacientes, tanto agudo de ángulo cerrado por desplazamiento anterior del complejo iris-cristalino o por la formación de sinequias posteriores y sinequias anteriores periféricas, o glaucoma crónico de ángulo abierto por el uso prolongado de glucocorticoides; membranas neovasculares coroideas en el 5-10% de los casos (sobre todo en los niños), fibrosis subretiniana y atrofia retinocoroidea extensa.
TratamientoNo existe una pauta establecida a seguir. Se han utilizado diferentes fármacos en el tratamiento del VKH recidivante, con eficacia variable.
Los glucocorticoides sistémicos son el fármaco de elección en los brotes agudos. La dosis inicial, la duración del tratamiento y la pauta descendente deben individualizarse en cada paciente11.
También se han empleado varios fármacos como los inmunosupresores, los fármacos biológicos y las inmunoglobulinas.
GlucocorticoidesSe emplean fundamentalmente en la fase aguda para evitar la disminución de la agudeza visual. La administración temprana de glucocorticoides puede prevenir la progresión de la enfermedad, acortar su duración y disminuir la afectación sistémica.
En casos graves, como en el desprendimiento seroso de retina, la administración inicial puede ser en forma de pulsos de metilprednisolona de 500-1.000mg/día durante 3 días consecutivos. Luego se continúa con la dosis habitual de 1mg/kg/día de prednisona oral, reduciendo la dosis de forma muy gradual, disminuyendo aproximadamente el 25% cada 2 semanas hasta alcanzar una dosis de 20 a 30mg/día; posteriormente se va disminuyendo la dosis a intervalos de 4 semanas hasta completar de 6 a 12 meses de tratamiento. En la fase de pauta descendente es cuando el paciente puede presentar una recaída con nuevo brote11,12.
En casos seleccionados pueden utilizarse glucocorticoides locales como la triamcinolona intravítrea. En pacientes con uveítis posterior unilateral refractaria está indicada la colocación de un implante intravítreo de fluocinolona que libera glucocorticoides durante 2,5años12,13.
InmunosupresoresDiferentes fármacos han sido utilizados en el tratamiento del síndrome VKH recidivante con eficacia variable, entre ellos la ciclosporina A, el clorambucilo, la ciclofosfamida, el tacrolimus, el micofenolato, la azatioprina, el metotrexato e incluso las inmunoglobulinas intravenosas.
Ciclosporina AEs el fármaco inmunosupresor de elección. La dosis indicada es de 5mg/kg/día. Está indicado en casos graves de uveítis posterior que no responden, o lo hacen débilmente, al tratamiento convencional.
AzatioprinaLa terapia con dosis bajas de azatioprina puede ser eficaz, con pocos efectos adversos, como ahorrador de glucocorticoides, especialmente en casos de uveítis resistentes a estos14. El tratamiento con 1,5mg/kg/día se asocia con la resolución de la neovascularización coroidea endógena14,15.
Algunos estudios han demostrado que los esteroides, cuando se utiliza en combinación con azatioprina o ciclosporina en uveítis grave, dan un mejor control de la inflamación, reduciendo así los efectos adversos de los glucocorticoides14,16.
La combinación de prednisona 40mg/día +azatioprina 1,5mg/kg/día +ciclosporina 5mg/kg/día lleva a los pacientes a una rápida resolución de la uveítis y a la resolución del desprendimiento de retina, con una mejoría en la agudeza visual más rápida.
Debido al efecto sinérgico de los fármacos, las dosis pueden ser reducidas rápidamente a dosis de mantenimiento sin reactivación de la enfermedad16.
Interferón alfaSe ha utilizado en casos esporádicos. Es una alternativa prometedora en formas corticorresistentes o corticodependientes de la enfermedad, pero son necesarios más estudios dirigidos a demostrar la utilidad del interferón alfa como tratamiento del VKH17.
AdalimumabLa dosis de 40mg/15días, de manera continuada durante más de 8 meses, parece ser una terapia eficaz y segura para el tratamiento de la uveítis refractaria y puede proporcionar la posibilidad de reducir los medicamentos inmunosupresores concomitantes en estos pacientes18. Actualmente hay varios estudios dedicados a evaluar el papel del adalimumab en el tratamiento de las uveítis.
InfliximabSe ha mostrado útil en el tratamiento de uveítis posteriores refractarias al tratamiento inmunomodulador convencional. Hay varios ensayos destinados a demostrar la eficacia del uso de infliximab como tratamiento de fondo de las uveítis, sobre todo en niños, al tener peor pronóstico, en dosis de 10-20mg/kg cada 6semanas19.
También existen estudios que apoyan el uso combinado de infliximab a 5mg/kg y metotrexato a 15mg/semana para minimizar la formación de anticuerpos anti-infliximab20,21.
EtanerceptExisten pocas publicaciones al respecto. Se ha descrito un caso donde se utilizó etanercept en un paciente diabético con buenos resultados, así como en niños con uveítis resistentes22.
RituximabLa experiencia de su utilización en uveítis se limita a casos puntuales. Se considera una alternativa terapéutica en pacientes que no responden al tratamiento con anti-TNF.
Se ha descrito un caso de un paciente refractario a tratamiento con altas dosis de glucocorticoides, metotrexato, ciclosporina, adalimumab y ranibizumab intravítreo, al que se le administraron 4 dosis de 1g de rituximab, logrando la remisión total después de 34 meses de seguimiento y preservando una buena agudeza visual23.
Fármacos biológicos intravítreos inhibidores del factor vascular de crecimiento endotelialTras la uveítis pueden producirse 2 complicaciones graves en la retina: el edema macular quístico y la neovascularización subretiniana (NVSR).
En ambos casos existe una alteración de la permeabilidad vascular que induce edema retiniano que, en el caso de la NVSR, se asocia al crecimiento de nuevos vasos sanguíneos desarrollando una membrana neovascular. Los fármacos intraoculares inhibidores del factor vascular de crecimiento endotelial cuentan principalmente con 2 efectos beneficiosos: la disminución de la permeabilidad vascular y la inhibición de la neovascularización patológica.
Los fármacos de que disponemos en la actualidad son bevacizumab, pegaptanib y ranibizumab. Existen publicaciones que confirman la eficacia de estos agentes frente al edema macular quístico secundarios a uveítis, por lo que deben tenerse en consideración al plantear una estrategia terapéutica24,25.
ConclusionesEl síndrome de VKH afecta a múltiples órganos, entre los que se incluyen el sistema ocular, el sistema nervioso, el auditivo y la piel. Etiológicamente es desconocido, aunque se cree que hay una reacción de autoinmunidad contra los melanocitos uveales y cutáneos. El diagnóstico es clínico cumpliendo una serie de criterios ya establecidos, aunque la angiografía fluoresceínica, la tomografía de coherencia óptica y la punción lumbar pueden ser de gran ayuda tanto para el diagnóstico como para valorar posibles complicaciones. El tratamiento de elección son los glucocorticoides sistémicos, pero en casos refractarios se suele usar tratamiento inmunosupresor. El pronóstico suele ser bueno, con más del 50% de pacientes con una agudeza visual final mayor a 20/50.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.