


Los fibroblastos sinoviales (FS) o sinoviocitos de tipo fibroblástico son el componente celular residente más importante de la membrana sinovial articular. Numerosos estudios apoyan la hipótesis de que los FS desempeñan una función relevante en la patogenia de la artritis reumatoide (AR). En la membrana sinovial reumatoide, los FS aumentan en número (hiperplasia) y muestran un fenotipo alterado que persiste en cultivo en ausencia de estímulos externos. Estas alteraciones se asocian a la activación de determinadas vías de señalización intracelular, que promueven el crecimiento celular y la expresión de múltiples factores como citocinas, quimiocinas, factores de crecimiento, moléculas de adhesión o enzimas de degradación de la matriz. La activación y expansión de los FS puede contribuir a procesos de reclutamiento, retención y activación de células inflamatorias, formación de nuevos vasos (angiogénesis), y destrucción del hueso y del cartílago. La contribución relativa de los FS a estos procesos es muy importante en los modelos animales, pero no se ha podido determinar en la AR humana donde no es posible intervenir terapéuticamente de manera específica sobre estas células. La identificación de las vías moleculares implicadas en las alteraciones de los FS reumatoides y su contribución fisiopatológica es la base para el desarrollo de terapias alternativas de la inflamación crónica y daño articular no dirigidas al sistema inmunitario.
Synovial fibroblasts (SF) or fibroblast-like synoviocytes are the major resident cellular component of joint synovial membrane. Numerous studies support the hypothesis that SF play an important role in the pathogenesis of rheumatoid arthritis (RA). In the RA synovial membrane, SF increase in number (hyperplasia) and exhibit an altered phenotype that persists in culture in the absence of external stimuli. These abnormalities are associated with the activation of specific signalling pathways that promote cell growth and the expression of multiple factors such as cytokines, chemokines, growth factors, adhesion molecules, and matrix degradation enzymes. The activation and expansion of SF appear to contribute to the recruitment, retention and activation of inflammatory cells, new blood vessel formation (angiogenesis), and bone and cartilage destruction. The relative contribution of SF to these processes is very important in animal models but has not been determined in human RA due to the lack of treatment interventions specifically targeting these cells. The identification of the molecular pathways involved in the altered phenotype of rheumatoid SF and their pathophysiological contribution are the basis for the development of new therapeutic alternatives for chronic inflammation and joint damage not targeting the immune system.
La artritis reumatoide (AR) es una enfermedad inflamatoria crónica de mecanismo autoinmune caracterizada por la presencia de autoanticuerpos (factor reumatoide y antipéptidos citrulinados) que preceden a la enfermedad, pero cuyo papel patogénico no ha sido completamente aclarado1. La enfermedad parece causada por numerosas influencias genéticas, comunes en parte a otras enfermedades autoinmunes, y relacionadas mayoritariamente con factores propios del sistema inmunitario adaptativo, con la concurrencia de algunos agentes ambientales como el humo del tabaco2,3.
Aunque se trata de un proceso de autoinmunidad sistémico, el desarrollo de la AR se localiza principalmente en las articulaciones verdaderas o diartrodiales y en las vainas tendinosas que comparten una estructura conjuntiva específica denominada membrana sinovial. El proceso inflamatorio se localiza en esta membrana y produce una destrucción de los elementos adyacentes, hueso, cartílago y tendones produciendo una severa pérdida funcional.
La membrana sinovial normal es una estructura especializada que tapiza la cavidad sinovial de las articulaciones diartrodiales y que elabora el líquido sinovial que lubrica y nutre al cartílago. La membrana sinovial está dividida en 2 capas: una capa íntima superficial limitante o lining, de una o 2 capas celulares de grosor, y una capa más profunda y menos definida de tejido conjuntivo vascularizado e inervado denominada sublining. El lining está formado fundamentalmente por 2 tipos celulares denominados sinoviocitos de tipo A o macrófagos sinoviales, y de tipo B o fibroblastos sinoviales (FS)4. Los macrófagos sinoviales son células fagocíticas con características similares a los macrófagos tisulares residentes en otros tejidos conjuntivos. Los FS tienen funciones sintéticas relacionadas con la homeostasis de la matriz extracelular y la secreción de componentes esenciales del líquido sinovial5.
En la fisiopatología de la AR participan todas las células responsables de la respuesta inmunitaria innata y adaptativa y las células residentes en la membrana sinovial objeto de esta revisión. La membrana sinovial reumatoide experimenta una serie de transformaciones que combinan la infiltración progresiva por células mononucleares reclutadas de la circulación junto a los cambios en las células residentes. En estas últimas, los cambios morfológicamente más evidentes son una transformación y crecimiento de la microvasculatura (angiogénesis), y un incremento de la celularidad de la zona limitante o del lining sinovial (hiperplasia). Así, la membrana sinovial se transforma en una masa de tejido creciente que se puede superponer e invadir la superficie del cartílago y del hueso (pannus)6. Este pannus se comporta como un tumor localmente invasivo cuyos componentes, macrófagos, osteoclastos y FS degradan hueso y cartílago7.
La formación de osteoclastos activos (osteoclastogénesis) es un proceso crítico en la erosión del hueso, dependiente de múltiples factores moleculares como M-CSF (del inglés macrophage colony stimulating factor) y el receptor activador de NFκB, el factor nuclear potenciador de las cadenas ligeras kappa de las células B activadas (RANKL), sobre el que influyen indirectamente numerosas citocinas como TNF-α (del inglés tumor necrosis factor-alpha). Las células capaces de promover la osteoclastogénesis desde sus precursores son los linfocitos T y los FS que expresan el RANKL. Por otra parte, los FS se han considerado los principales efectores de la destrucción del cartílago por su capacidad de invasión y producción de colagenasas y otras proteasas de la matriz8.
Por tanto, aunque la patogenia de la AR implica una respuesta autoinmune, una vez establecida la enfermedad el proceso local es muy complejo y no exclusivamente dependiente de la respuesta inmunitaria. Desde hace décadas se investiga la contribución de los FS en la iniciación, perpetuación o destrucción articular en la AR, con la expectativa de desarrollar agentes terapéuticos alternativos a los actuales, dirigidos a factores o células del sistema inmunitario y con efectos inevitables sobre los mecanismos naturales de defensa.
Fibroblasto sinovialLos fibroblastos son células de origen mesenquimatoso residentes en el tejido conjuntivo que sintetizan los componentes de la matriz extracelular necesarios para la homeostasis o la reparación de lesiones. Se suelen identificar indirectamente por su morfología en forma de huso, su capacidad de adherencia al plástico en cultivo y la ausencia de marcadores leucocitarios (CD45-negativos) o de otras estirpes celulares. Son morfológica y funcionalmente heterogéneos, con diversos fenotipos dependiendo de su localización y del estado tisular9. El patrón de expresión genética específico de cada tejido podría depender de mecanismos epigenéticos de regulación. Estos mecanismos parecen alterarse en las enfermedades inflamatorias, y ello podría explicar los diferentes cambios fenotípicos observados en los fibroblastos procedentes de distintas lesiones inflamatorias.
Los fibroblastos son células metabólicamente activas y capaces de desarrollar características específicas en respuesta al microambiente. Los fibroblastos de diferentes tejidos cutáneos, viscerales o sinoviales muestran patrones diferentes de expresión génica, diferencias que se acentúan en respuesta a estímulos10. Los FS y dérmicos tienen patrones basales similares de expresión génica, pero divergen después de su estimulación con TNF-α, produciendo diferentes cantidades de IL-6, IL-8 y de la proteína quimioatrayente de monocitos (MCP-1)/CCL2, y estas diferencias parecen funcionalmente importantes11.
Es posible que los denominados FS incluyan una población celular heterogénea en términos de origen, localización y fisiología. De forma simplificada, los FS parecen proceder de células pericondriales que durante el desarrollo fetal expresan altos niveles del receptor del hialuronato CD44, y que forman la interzona que dará lugar a la cavidad articular, una región de células aplanadas y agrupadas en capas que producen matriz extracelular y colágenos tipo i y iii4. Estas células progenitoras también dan lugar a las células estromales de la médula ósea con las que los FS reumatoides tienen además de este origen común, diversas similitudes funcionales como la capacidad de producir la quimiocina CXCL12/SDF-1 (del inglés stromal cell-derived factor-1) o la molécula de adhesión VCAM-1 (del inglés vascular cell adhesion molecule-1), críticas en el mantenimiento del nicho hematopoyético. Estos factores pueden cobrar especial relevancia en la inflamación sinovial, donde pueden ejercer funciones similares de retención y soporte a las células mieloides y linfoides que infiltran la sinovial inflamada.
Existen también similitudes en el desarrollo del estroma de los órganos linfoides secundarios y el estroma sinovial, que durante la inflamación expresa quimiocinas asociadas al homing linfoide como CXCL13, CCL19 y CCL21, y ejerce funciones de células reticulares o foliculares dendríticas, constitutivamente presentes solo en órganos linfoides12,13. La persistencia de progenitores estromales (MSC, mesenchymal stem cells) capaces de rediferenciarse en otras poblaciones celulares como osteoblastos o condrocitos, es otra característica común al estroma medular y sinovial14. Algunas vías potencialmente relacionadas con el desarrollo embrionario y el fenotipo de los FS y las MSC se han relacionado con el sistema Wnt y sus ligandos15.
La función principal de los FS en la articulación sinovial normal es contribuir a la homeostasis de la articulación, produciendo los componentes de la matriz extracelular y del líquido sinovial. En la membrana sinovial, los FS ocupan 2 posiciones o subpoblaciones, con diferentes características morfológicas y biológicas. Unos forman la capa íntima o lining, formando una estructura mesotelial que limita la cavidad sinovial, y otros forman parte de la subíntima o sublining, similares a los fibroblastos intersticiales de otros tejidos. Los FS del lining expresan marcadores específicos como la enzima uridina difosfato glucosa deshidrogenasa (UDPGD), las moléculas de adhesión VCAM-1 y cadherina-11 o el factor del complemento CD55 (DAF, decay-accelerating factor)4. Estos marcadores definen funciones específicas de los FS del lining como la producción de elevadas cantidades de ácido hialurónico debida a la alta actividad de la enzima UDPGD16. En modelos animales deficientes en la molécula de adhesión homotípica cadherina-11 se ha demostrado que esta es esencial en la formación y organización seudoepitelial del lining, y en la destrucción del cartílago durante la artritis, pero no del hueso17.
Tanto los FS del lining como los del sublining pueden interaccionar con las células inflamatorias mieloides o linfoides mediante moléculas de adhesión como VCAM-1 o a través de la producción de numerosos factores paracrinos como citocinas y quimiocinas relacionadas con el reclutamiento, migración, activación o supervivencia de la mismas. Esta capacidad de las células del estroma de influir en otras poblaciones se ha demostrado en la médula ósea y los tejidos linfoides normales donde constituyen el nicho de las células hematopoyéticas o del sistema inmunitario, y parece también relevante para el mantenimiento crónico de la infiltración celular en la sinovial12,13.
Estudio de los fibroblastos sinovialesLos procedimientos habituales de estudio de los FS incluyen la histopatología del tejido sinovial acompañada de técnicas de estudio de expresión molecular y su expansión y cultivo en placas de plástico. El estudio de las características y comportamiento de los FS in vitro ha generado abundante información sobre el fenotipo de los FS en la AR. Los FS en cultivo proliferan con facilidad y pueden mantener algunas características durante múltiples generaciones. Sin embargo, no está claro si el cultivo de los FS representa una mezcla heterogénea de las poblaciones fibroblásticas originalmente presentes en la membrana sinovial (fibroblastos de lining o sublining, MSC y pericitos) y en qué proporción, o si existe una expansión preferente de determinadas subpoblaciones a través de la proliferación18,19.
La proliferación forzada en cultivos monocapa sobre plástico también es un factor importante no presente en el medio sinovial, donde estas células proliferan lentamente y viven en una matriz tridimensional. Por lo tanto, el estudio de los FS en cultivo presenta ciertas limitaciones, y aunque permite analizar diferencias entre grupos estas deben ser confirmadas en estudios tisulares.
La mayor parte de la investigación sobre los FS se ha realizado en la AR, existiendo poca información en otras enfermedades articulares. Los tejidos y cultivos tradicionalmente se han obtenido durante la cirugía de sustitución protésica, una situación terminal alejada de las fases iniciales del proceso artrítico. Los FS reumatoides han sido generalmente comparados con los artrósicos, usados como control, mientras que muy pocos estudios incluyen FS de sinovial normal, cuestionando en gran medida la interpretación de los resultados obtenidos en AR. Los FS artrósicos pueden expresar citocinas inflamatorias o enzimas que degradan la matriz de manera anormal, en algunos estudios en menor grado que los reumatoides, pero no pueden ser considerados «normales»20. Un reciente estudio sistemático de FS de pacientes con artrosis, AR y controles sanos pone de manifiesto esta importante limitación demostrando importantes cambios transcriptómicos en los FS procedentes de articulaciones artrósicas comparadas con las de individuos sanos21.
Otros estudios han demostrado alteraciones cromosómicas similares en los FS de pacientes con AR, artrosis y algunas espondiloartropatías, comparados con FS de donantes sanos. Estas modificaciones incluyen regiones que contienen genes de citocinas, factores de crecimiento, moléculas de señalización, factores de transcripción y proteasas, pero su significado y consecuencias funcionales no se conocen22.
En otras enfermedades causantes de artritis crónica como la artritis psoriásica y otras espondiloartropatías se han realizado menos estudios que en general sugieren que los FS también sufren cambios similares a los observados en la AR, como la hiperplasia o la expresión de proteasas de la matriz23–25. Por tanto, la mayoría de las alteraciones de los FS que se describen a continuación corresponden a la AR.
El fibroblasto sinovial en la artritis reumatoideCrecimiento de los fibroblastos sinoviales en la artritis reumatoideUna de las observaciones histológicas clásicas en la AR es la expansión de los fibroblastos de la capa del lining sinovial, definida como hiperplasia y ligada tradicionalmente al crecimiento de la membrana sinovial y a la formación del pannus26,27. Esta descripción morfológica de un aumento en el número de capas del lining en tejidos reumatoides corresponde en realidad a una acumulación de macrófagos y fibroblastos4. Sobre la población de FS del sublining hay pocos datos cuantitativos debido a la falta de marcadores adecuados, ya que los marcadores conocidos como VCAM-1, cadherina-11 o CD55 son principalmente expresados por los FS del lining. Por otra parte, estos marcadores son inducibles en FS estimulados con TNF-α, presente en la sinovial reumatoide, y podrían reflejar también un estado de activación además de la localización o cantidad de estos FS28–30.
Recientemente mediante la detección inmunohistoquímica de una chaperona específica de colágeno (hsp47) universalmente expresada por fibroblastos se ha podido estudiar la evolución de ambas subpoblaciones en el curso de la AR. La expansión de los fibroblastos del lining se incrementa paralelamente a la actividad y a la progresión de la enfermedad, mientras que la hiperplasia de los fibroblastos del sublining se comporta de forma opuesta31. Asimismo, la hiperplasia fibroblástica del lining disminuye significativamente tras la terapia con antagonistas del TNF-α, aunque no se normaliza completamente ni siquiera en pacientes en remisión. Esta hiperplasia de los FS junto a una expansión vascular persistente tras el tratamiento podrían contribuir a la progresión subclínica y reactivación de la enfermedad en pacientes que han alcanzado la remisión clínica31,32.
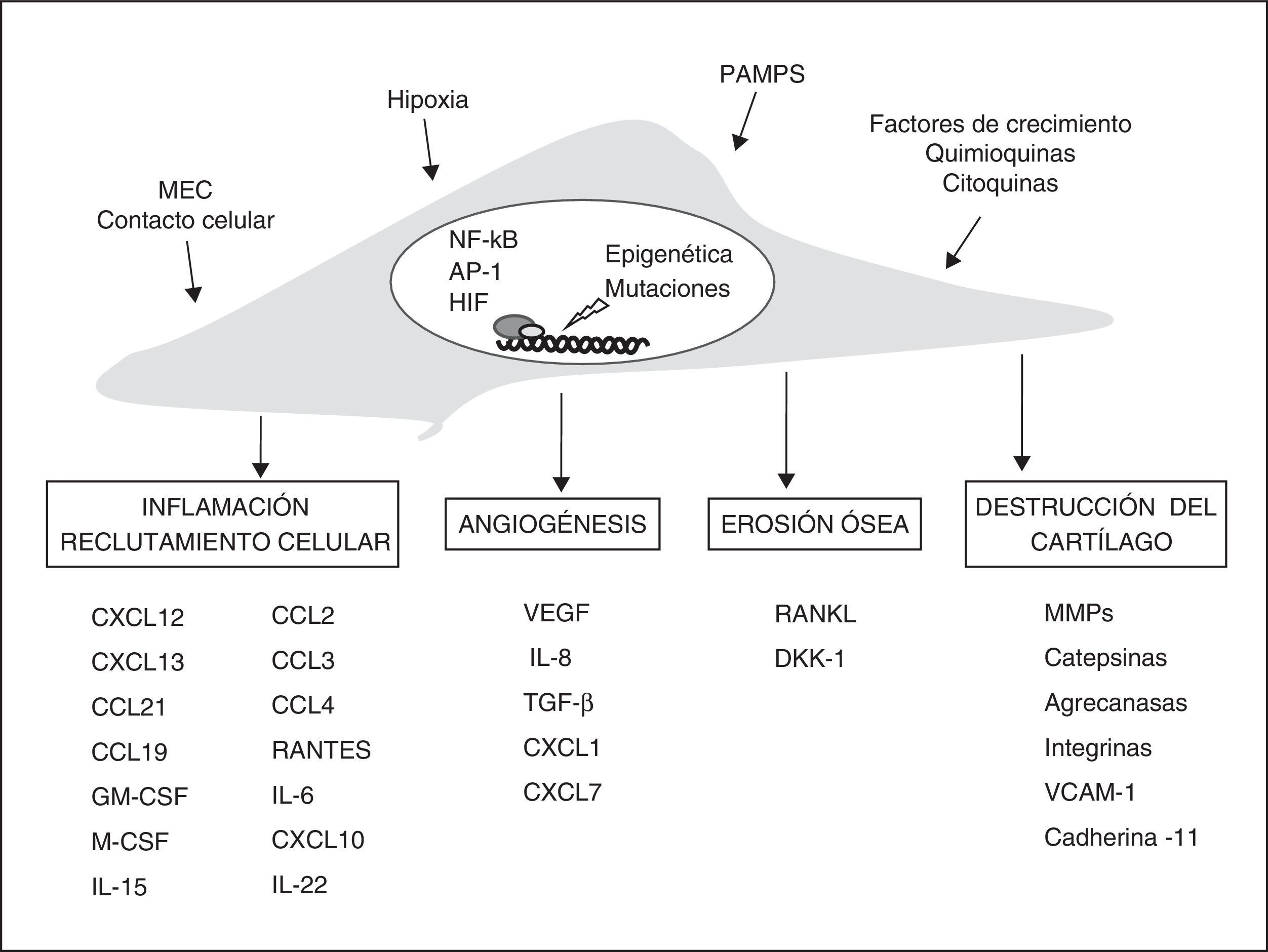
Fenotipo del fibroblasto sinovial de la artritis reumatoideEn la AR, diferentes estudios apoyan la hipótesis de que los FS desempeñan un papel central en el daño a la articulación y en la persistencia de la inflamación crónica. Se ha sugerido que los FS reumatoides muestran un fenotipo comparable en determinados aspectos al de algunas células transformadas. Pueden crecer sin adherirse y escapar a la inhibición por contacto como las células tumorales33. Los FS reumatoides son también más resistentes a la muerte celular por apoptosis34. Además, responden a las citocinas proinflamatorias aumentando su crecimiento y resistencia a la muerte y produciendo numerosos mediadores inflamatorios y de destrucción tisular (fig. 1). Este fenotipo es inicialmente una respuesta a estímulos producidos en el ambiente proinflamatorio que puede después perpetuarse de manera autónoma.

Vías de participación de los fibroblastos sinoviales en la AR. Estímulos y cambios intracelulares más importantes modificadores del fenotipo de los FS y sus mecanismos efectores en la artritis crónica. AP-1: proteína activadora; DKK-1: Dickkopf-1; GM-CSF y M-CSF: factores de estimulación de colonias de granulocitos y macrófagos; HIF: factor inducible por hipoxia; MEC: matriz extracelular; MMPS: metaloproteinasas; NFκB: factor nuclear potenciador de las cadenas ligeras kappa de las células B activadas; PAMPS: patrones moleculares asociados a patógenos; RANKL: receptor activador de NFκB; TGF-β: factor de crecimiento transformante-β; VCAM-1: molécula de adhesión celular vascular-1; VEGF: factor de crecimiento endotelial vascular.
Entre los diversos estímulos que activan a los FS, se incluye la activación de los receptores de tipo Toll (TLR, Toll-like receptors), los receptores de citocinas y la unión de integrinas de membrana a la matriz extracelular. Los efectos de las citocinas macrofágicas (TNF-α y IL-1β) sobre los FS producen una potente respuesta efectora que incluye la inducción de otras citocinas como IL-6, quimiocinas, y proteasas de la matriz extracelular. La estimulación con TNF-α produce cambios epigenéticos en los FS que permiten la prolongación en el tiempo de su estado de activación en ausencia del estímulo inicial, explicando en parte la persistencia de la respuesta proinflamatoria35. Además, la respuesta de los FS a TNF-α parece necesaria en la inflamación crónica articular, de manera que los ratones con fibroblastos deficientes en receptores de TNF-α de tipo i no desarrollan artritis en respuesta al TNF-α36.
Estudios ex vivo de FS reumatoides confirman su fenotipo permanentemente activado. Su implantación subcutánea en ratones inmunodeficientes y en ausencia de estímulos adicionales produce una potente respuesta inflamatoria marcada por el reclutamiento de células mieloides y por la angiogénesis37. En un modelo similar los FS reumatoides, pero no los artrósicos, coimplantados junto a cartílago humano producen su destrucción progresiva38. En este modelo experimental los FS de la AR, pero no los artrósicos o los sanos, son también capaces de migrar de unas articulaciones a otras propagando la enfermedad39.
Alteraciones genéticas y epigenéticas de los fibroblastos sinoviales en la artritis reumatoideDiversos estudios han intentado explicar esta alteración del fenotipo de los FS reumatoides, bien por mutaciones en distintos genes, bien por modificaciones epigenéticas. Los FS reumatoides tienen una expresión incrementada de oncogenes (c-myc, c-fos) y en algunos pacientes se han descrito mutaciones en genes reguladores del ciclo celular, proliferación y apoptosis, otra similitud con las células tumorales5. La mutación mejor caracterizada es la del gen supresor de tumores p53 que podría contribuir a las alteraciones del crecimiento y fenotipo observadas en los FS40. La pérdida de función de p53 en los FS disminuye la tasa de apoptosis y aumenta la expresión de citocinas y, en el modelo de artritis inducida por colágeno, conduce a una mayor severidad y destrucción de la articulación41. Sin embargo, otros estudios han observado que estas mutaciones son infrecuentes en FS, aunque la expresión de p53 funcional puede desempeñar un papel patogénico no relacionado con la supresión del crecimiento en la AR o sus modelos experimentales42,43.
La expresión génica también puede ser regulada de forma estable en el tiempo por mecanismos epigenéticos como los cambios en la metilación del ADN y la acetilación de histonas. Estas modificaciones facilitan o impiden el acceso de los factores necesarios para la transcripción de los genes. Los cambios epigenéticos pueden ser revertidos y están regulados por enzimas específicas.
Desde un punto de vista global se ha observado que en los FS de la AR hay una reducción generalizada de la metilación del ADN y de la acetilación de histonas. Los cambios en los patrones de metilación son heredados durante la proliferación por las células hijas manteniendo el fenotipo en sucesivas generaciones celulares. La hipometilación global de los FS de la AR podría deberse a una menor expresión de la enzima DNA metiltransferasa 1 (DNMT1) y un aumento en enzimas que intervienen en el reciclado de poliaminas44,45. La hipometilación de los FS reumatoides podría ser responsable de la activación de múltiples genes entre los que se han descrito secuencias repetitivas normalmente silenciadas, como los elementos retrovirales endógenos LINE-1, secuencias Alu y microsatélites46. También se ha descrito una demetilación en el promotor del gen de la IL-6 que incrementa su expresión47. Otra prueba indirecta de la importancia de este mecanismo es la observación de un fenotipo similar al de los FS reumatoides en FS normales cultivados en un ambiente hipometilante48. Además, las citocinas proinflamatorias de la AR (TNF, IL-1β e IL-6) inducen hipometilación del ADN49,50. También se ha descrito la hipermetilación del promotor del gen de receptor de muerte 3 (DDR3), asociada a su menor expresión y a una mayor resistencia a la apoptosis en los FS de la AR51.
Las modificaciones de histonas incluyen mecanismos de acetilación, metilación y sumoilación. Se ha descrito un aumento en los niveles de las enzimas histona desacetilasas (HDAC), HDAC1 y sirtuína i en los FS reumatoides, y además el TNF-α incrementa esta expresión52,53. La sobreexpresión de estas HDAC aumenta la expresión de citocinas proinflamatorias y la proliferación celular e inhibe la apoptosis en los FS. Diversos estudios investigan el efecto de inhibidores de las HDAC como el MI192, la tricostatina A (TSA) o la nicotinamida54. Estos inhibidores disminuyen la expresión de citocinas proinflamatorias y además se ha observado una mejora de las manifestaciones clínicas en ratones con artritis inducida por anticuerpos de colágeno tratados con TSA55. Sin embargo, los cambios en los niveles de acetilación/desacetilación ocurren también en proteínas no histonas y por lo tanto los efectos observados podrían también deberse a otros mecanismos.
Otra importante vía de regulación de la expresión génica son los microARN (miARN), pequeños ARN no codificantes que regulan los niveles y reducen la traducción proteica de múltiples ARN mensajeros (ARNm). Los miARN se han implicado en la regulación de procesos importantes en la AR como la proliferación, diferenciación y muerte celular. Los FS reumatoides tienen constitutivamente aumentados los niveles de miR-146a y miR-155 comparados con los artrósicos, y la expresión de estos miARN aumenta en respuesta a la estimulación por citocinas o agonistas de los TLR. Se ha demostrado que la inducción de miR-155 reduce específicamente la expresión de las MMP-1 y 3, y podría por tanto contribuir a modular la destrucción articular56.
Aunque en conjunto estos cambios son complejos y su conocimiento es aún limitado, las observaciones mencionadas apoyan la hipótesis de que las alteraciones epigenéticas explicarían la heredabilidad y persistencia del fenotipo de los FS reumatoides.
El fibroblasto sinovial como mediador de daño articularLos FS de la AR desempeñan un papel fundamental en la destrucción de cartílago y hueso articular. Actúan directamente degradando la matriz del cartílago, e indirectamente sobre el hueso como inductores de la diferenciación y activación de los osteoclastos.
La degradación del cartílago es un proceso en varias etapas, donde interviene la adhesión de los FS al cartílago y la síntesis de enzimas extracelulares o de membrana que degradan la matriz extracelular compuesta por colágeno y proteoglucanos. Las moléculas de adhesión, particularmente las integrinas, facilitan el anclaje de los fibroblastos a la matriz del cartílago. Los fibroblastos en cultivo expresan abundantemente varias integrinas β157. In vitro, es posible inhibir parcialmente la unión de los fibroblastos al cartílago mediante anticuerpos anti-β1. Además, esta unión a integrinas modula señales intracelulares en los FS relevantes en la patogenia de la AR, incluyendo MAPK (mitogen-activated protein kinases) y Ras, que también contribuyen a inducir la expresión de metaloproteinasas de matriz (MMP)58. Otras moléculas de adhesión como VCAM-1 también parecen contribuir a la invasión del cartílago38. Este proceso de adhesión e invasión-degradación del cartílago puede reproducirse in vitro mediante cocultivo de FS reumatoides y cartílago normal en ausencia de estímulos. In vivo, en ratones inmunodeficientes, los implantes de FS reumatoides sobre injertos de cartílago humano sano se adhieren al cartílago y lo invaden espontáneamente, aunque el proceso puede ser también estimulado por citocinas exógenas como TNF-α e IL-1β59. La formación de un lining hiperplásico mediante la agregación homotípica de FS parece ser relevante en la degradación del cartílago en el modelo de ratones deficientes en cadherina-11, donde la alteración de este proceso previene el daño al cartílago durante la artritis inducida por colágeno17.
Entre los efectores de la destrucción del cartílago producidos por los FS reumatoides destacan las proteasas MMPs, catepsinas y agrecanasas. Entre las MMPs, las colagenasas (MMP-1 y -13) y la estromelisina (MMP-3) parecen especialmente importantes en la degradación del cartílago reumatoide60. Su síntesis y activación es inducida por varios factores, incluyendo citocinas proinflamatorias (IL-1β, TNF-α), factores de crecimiento, ligandos de TLR o especies reactivas de oxígeno59 (fig. 1). Los FS en cultivo también producen constitutivamente las proteínas TIMP (tissue inhibitors of MMPs), que bloquean específicamente las MMP61. Las catepsinas son proteasas de amplia especificidad y son también reguladas por citocinas y protooncogenes62. Las agrecanasas son también importantes mediadores de la destrucción cartilaginosa y algunas son expresadas constitutivamente por los FS en cultivo (ADAMTS-4 y 5)63.
La erosión ósea es necesariamente mediada por osteoclastos, cuya diferenciación está fundamentalmente regulada por el sistema RANK/RANKL (receptor activador de NF-кB y su ligando)64. Los ratones deficientes en RANKL no pueden formar osteoclastos, pero sí desarrollar sinovitis. La inducción de artritis en estos ratones produce inflamación articular y degradación del cartílago pero no pérdida ósea65. Los precursores osteoclásticos responden a la activación del receptor RANK por su ligando específico RANKL, transformándose en osteoclastos multinucleados capaces de degradar el hueso (osteoclestogénesis). El RANKL es exclusivamente expresado en el tejido sinovial por LT activados y FS. Los estudios histológicos sobre la interfase sinovial de las erosiones reumatoides indican que la expresión de RANKL por los FS del pannus es crítica en la erosión66. Además, los FS de la AR incrementan la expresión del inhibidor osteoblástico Dickkopf-1 (DKK-1), bloqueando los mecanismos de reparación ósea67. Por tanto, los FS reumatoides contribuyen significativamente a la erosión ósea a través de múltiples mecanismos.
Fibroblasto sinovial y regulación de la inflamaciónLos FS reumatoides participan activamente en el reclutamiento y retención de las células del infiltrado inflamatorio mediante la secreción de citocinas y quimiocinas. Además, secretan diversos factores moleculares que favorecen la supervivencia y proliferación de las células reclutadas. En pacientes con AR se han detectado niveles elevados de numerosas quimiocinas y sus respectivos receptores. Los fibroblastos de la AR pueden producir quimiocinas en exceso de forma constitutiva o después de ser estimulados con citocinas proinflamatorias, micropartículas, hipoxia, ligandos de los TLR u otras quimiocinas (fig. 1).
Los FS producen tanto quimiocinas relacionadas con procesos homeostáticos de tráfico celular mieloide y linfoide (quimiocinas de homing medular y linfoide) como aquellas inducibles durante procesos inflamatorios (quimiocinas inflamatorias). Entre las primeras CXCL12/SDF-1 que se han relacionado con el reclutamiento y retención de las células T, particularmente de memoria, en la articulación inflamada68. Los FS sinoviales son también la fuente principal de las quimiocinas denominadas homeostáticas o de homing linfoide (CXCL13, CCL21, CCL19), normalmente solo presentes en órganos linfoides secundarios, y que se asocian a la formación de estructuras linfoides terciarias (neogénesis linfoide) en la sinovial reumatoide69,70.
Los FS estimulados con IL-1β o TNF-α producen numerosas quimiocinas inflamatorias, quimiotácticas para monocitos y macrófagos, contribuyendo al reclutamiento de estos tipos celulares a la articulación inflamada. Algunos ejemplos de estas quimiocinas son CCL5/RANTES (regulated on activation normal T cell expressed and secreted), la proteína inducible por interferón-10 (IP-10)/CXCL10, ENA-78, CCL2/MCP-1 y las proteínas inflamatorias de macrófagos MIP-1α/CCL3 y MIP-1β/CCL471. Múltiples quimiocinas pueden desempeñar otras funciones sobre los leucocitos y sobre otros tipos celulares, particularmente células endoteliales o precursores, y como se señala más adelante participan en el proceso de angiogénesis, muy activo en la sinovial reumatoide.
Los mediadores centrales del proceso inflamatorio reumatoide son las citocinas proinflamatorias macrofágicas, particularmente TNF-α, que no es sintetizado por los FS. Sin embargo, estos parecen la fuente más importante de IL-6 cuyo papel central también ha sido confirmado terapéuticamente72. Los fibroblastos de la AR en cultivo producen espontáneamente IL-6 y esta producción puede aumentar con la presencia de diversos estímulos, incluidas las citocinas IL-1β o TNF-α.
La citocina IL-15 es otra citocina de origen fibroblástico muy abundante en el líquido sinovial de pacientes con AR. Los fibroblastos en cultivo de estos pacientes producen constitutivamente elevados niveles de esta citocina73,74. En cocultivos de FS y linfocitos se ha demostrado la importancia de IL-15 en el diálogo entre ambos elementos celulares, contribuyendo finalmente a la supervivencia y activación linfocitaria75.
Los pacientes con AR también tienen mayores niveles de IL-22 comparados con los donantes sanos, niveles que se correlacionan con la actividad de la enfermedad y la presencia de erosiones óseas. En la AR, esta citocina se expresa principalmente en FS y macrófagos pero solo los FS expresan el receptor 1 de IL-22. Estudios recientes sugieren que IL-22 tiene efectos proinflamatorios y osteoclastogénicos en la AR, posiblemente por su papel sobre la proliferación y activación de los FS76,77. Otras citocinas descritas en la AR y producidas por fibroblastos estimulados son la IL-18, IL-33 e IL-3278.
Los factores de estimulación de colonias de granulocitos (GM-CSF) y de macrófagos (M-CSF) son también abundantes en el tejido y líquido sinovial de pacientes con AR y son liberados principalmente por las células del lining. La estimulación de los fibroblastos in vitro por IL-1β o TNF-α aumenta de forma importante la producción de GM-CSF que podría contribuir a la expansión local de los macrófagos79.
Los FS reumatoides en cultivo son una fuente importante de interferones (IFN) de tipo i que pueden tener distintas funciones inmunoactivadoras en esta enfermedad. Aunque en general tienen efectos proinflamatorios como la liberación de quimiocinas y MMP, en diversos modelos de artritis también se ha demostrado un efecto antiinflamatorio de IFN-β80. Los fibroblastos tienen también la capacidad de producir otras citocinas antiinflamatorias y factores potencialmente supresores de la sinovitis, como el factor de crecimiento transformante TGF-β, e IL-1RA, el antagonista natural de la IL-181,82.
Fibroblasto sinovial y angiogénesis en la artritis reumatoideEn la sinovial reumatoide, de manera similar a otras enfermedades inflamatorias crónicas, se ha descrito un aumento de la vascularización sinovial y de la expresión de factores proangiogénicos que reflejan un proceso neoangiogénico activo. Se postula que este proceso es necesario para mantener el metabolismo y la incorporación celular durante el proceso inflamatorio crónico83–85. Los FS de la AR producen múltiples mediadores proangiogénicos capaces de inducir el reclutamiento, la proliferación y la activación de las células endoteliales como el factor de crecimiento endotelial vascular (VEGF). Numerosas quimiocinas como IL-8, CXCL12, ENA-78, CXCL1, CXCL7, MCP-1 y fractalcina (CXC3CL1) también parecen ejercer efectos proangiogénicos directos o indirectos en la AR86,87. Los FS y macrófagos sinoviales sintetizan VEGF de forma abundante, a niveles detectables sistémicamente. El nivel plasmático de VEGF es un buen marcador sistémico de inflamación articular en la AR88. La marcada hipoxia de la sinovial reumatoide parece el factor más potente inductor de VEGF en los FS a través del factor de transcripción inducible por la hipoxia HIF-1α89. Otros mediadores como TNF-α, IL-1790,91, la activación del TLR292 y la unión de CD4093 pueden contribuir al proceso (fig. 1).
Los fibroblastos reumatoides implantados a ratones inmunodeficientes y en ausencia de estímulos adicionales inducen el reclutamiento de células mieloides y el desarrollo de vasos sanguíneos37. Un proceso similar se ha observado en el estroma tumoral, donde tiene importantes implicaciones en el crecimiento tumoral, que parecen también aplicables a la perpetuación de la inflamación crónica94–96. Esta capacidad de los FS reumatoides depende del eje HIF-1α, VEGF y CXCL12/CXCR4. Los factores VEGF y CXCL12 son dianas transcripcionales de HIF-1α en condiciones de hipoxia y representan un nexo adicional entre la hipoxia y la infiltración por células inflamatorias97,98. Tanto la inhibición de la actividad de HIF-1α como el uso de antagonistas de VEGF o CXCL12/CXCR4 pueden bloquear la capacidad angiogénica y de reclutamiento celular de los FS de la AR37.
Terapias dirigidas contra el fibroblasto sinovialLas dianas de las terapias actuales para la AR se dirigen fundamentalmente contra células T, células B o citocinas macrofágicas. Todas estas terapias disminuyen la actividad de la enfermedad y retrasan la destrucción articular. Sobre la membrana sinovial, todas ejercen efectos similares, reduciendo la infiltración celular linfoide y macrofágica en paralelo a la respuesta clínica70. También las células residentes sufren cambios en respuesta a las terapias, reduciéndose la hiperplasia fibroblástica del lining, pero no del sublining, y modificándose en parte la estructura vascular31,32. Sin embargo, la reducción de estos elementos es solo parcial, persistiendo una importante hiperplasia fibroblástica y vascular tras el tratamiento con anti-TNF-α31,32. Se desconoce el significado pronóstico de estos cambios residuales sobre el daño estructural o las futuras recidivas inflamatorias una vez alcanzada una mejoría clínicamente aceptable de la inflamación articular.
No existen terapias específicamente dirigidas contra los fibroblastos, pero sí frente a algunos de sus factores. La intervención directa sobre la IL-6 es muy eficaz en la AR pero esta citocina está también producida por otros tipos celulares. Los antagonistas de IL-15, otra citocina fibroblástica, también parecen tener algún efecto terapéutico en la AR, aunque no han alcanzado desarrollo clínico99. Los antagonistas de la señalización intracelular o los de NFκB o MAPK podrían actuar sobre procesos FS-dependientes, además de interferir con la función de otros muchos elementos celulares que contribuyen a la inflamación y respuesta autoinmune.
Las únicas intervenciones fibroblasto-específicas se han realizado en animales, con las limitaciones propias de estos modelos. Como se ha señalado, la interferencia con la formación del lining fibroblástico mediante la deleción de la molécula cadherina-11 previene en ratones el desarrollo de artritis y de daño cartilaginoso, aunque no óseo17. La deleción específica de los receptores TNFR1 en fibroblastos previene la inflamación articular mediada por TNF-α en ratones, y esto representa la prueba más importante de la importancia de los FS como las principales células efectoras de TNF-α en la artritis36.
Sin embargo, no existen pruebas directas de este concepto en las enfermedades inflamatorias articulares humanas, debido a la falta de terapias específicamente dirigidas a los FS. La investigación de factores involucrados en la artritis específicos de los FS es un paso intermedio necesario para hacer posibles estas intervenciones. Por tanto, la potencial seguridad y eficacia de tales intervenciones es aún incierta. El desarrollo de nuevas estrategias de intervención dirigidas a los FS permitiría el abordaje terapéutico de la artritis crónica desde una perspectiva diferente de la actual, esencialmente dirigida a los elementos del sistema inmunitario y cuyas limitaciones en eficacia y seguridad son bien conocidas.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.
Los autores han recibido ayudas a la investigación del Instituto de Salud Carlos III a través de la Red de Investigación en Inflamación y Enfermedades Reumáticas (RIER) RD12/09/016 y de la Comunidad Autónoma de Madrid a través de la red S2010/BMD-2350 (RAPHYME). Elena Izquierdo ha recibido apoyo del programa de formación posdoctoral Juan de la Cierva del Ministerio de Economía y Competitividad (MINECO).