



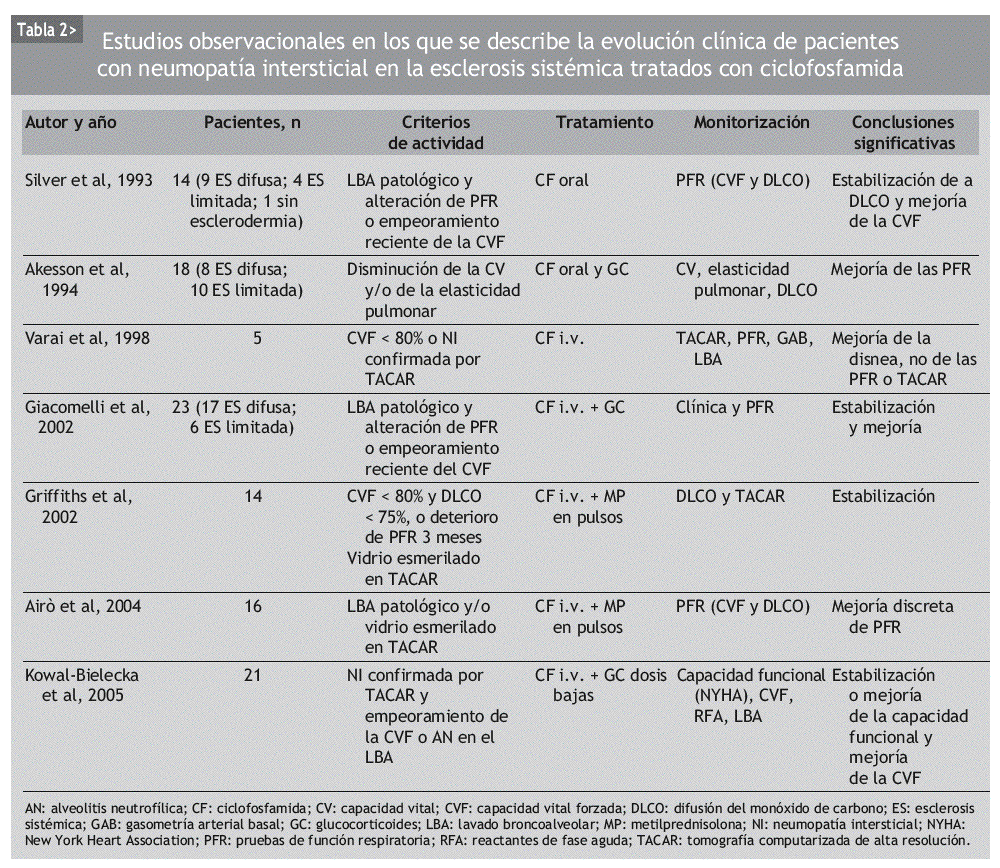
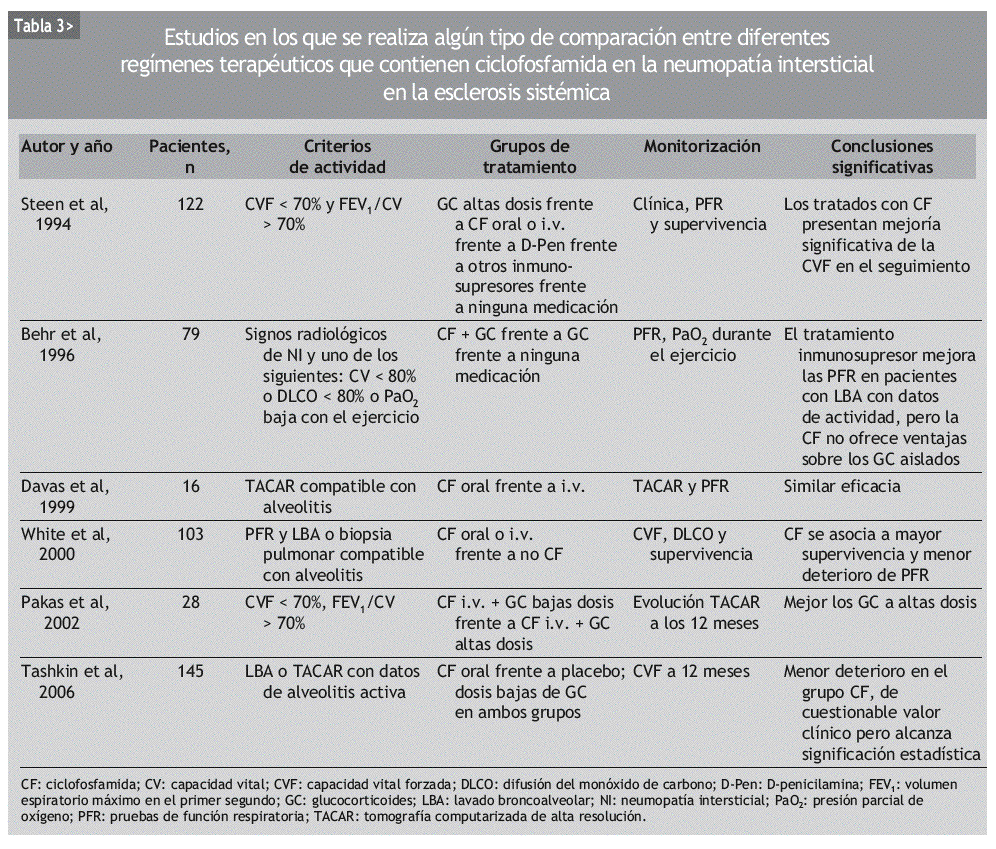
INTRODUCCION
La esclerosis sistémica (ES) es una enfermedad del tejido conectivo de etiología desconocida, caracterizada por la presencia de daño microvascular, fibrosis excesiva de la piel y afección de diversos órganos, como el corazón, los pulmones, el riñón o el tracto digestivo1. La neumopatía intersticial (NI) clínicamente significativa aparece en aproximadamente un 40% de los pacientes con ES. Con la aparición de los inhibidores de la enzima de conversión de la angiotensina, la NI ha sustituido a la nefropatía como la primera causa de morbilidad y mortalidad2,3. Ni la extensión ni la gravedad de la afección cutánea se correlacionan con la afección pulmonar. Aunque es la forma difusa de la ES lo que se asocia más frecuentemente a la NI, también aparece en la forma limitada de la ES y en las formas sin esclerodermia. Los estudios de autopsias revelan la existencia de una NI en un 70-100% de los pacientes con ES4.
FACTORES DE RIESGO
Aunque ninguno de los factores de riesgo encontrados predice con seguridad en qué pacientes con ES se desarrollará una NI, se ha considerado como tales el sexo masculino, la raza negra, la corta duración de la ES, los anticuerpos anti-Scl 70, una reducción temprana o progresiva de la capacidad vital forzada (CVF), la velocidad de sedimentación globular (VSG) elevada y la afección cardíaca o esquelética. Los anticuerpos anticentrómero se asocian a una menor prevalencia de fibrosis pulmonar. Por el momento, no se ha encontrado ninguna relación consistente con el tabaco3,5.
EPIDEMIOLOGIA
La prevalencia de la fibrosis pulmonar en la ES oscila del 25 al 90%, dependiendo de la raza de los pacientes estudiados y de los medios utilizados para identificarla. Si se utilizan las pruebas de función respiratoria (PFR) con una CVF menor del 80%, la prevalencia varía del 25 al 45%, mientras que si se usa la tomografía computarizada de alta resolución (TACAR), la prevalencia puede alcanzar el 90%. Aparece en pacientes con ES difusa y limitada, aunque más frecuentemente en la forma difusa6.
ANATOMIA PATOLOGICA
Hasta hace unos años, el conocimiento sobre la histopatología del pulmón en la ES provenía de estudios con autopsias, que tendían a resaltar los cambios fibróticos finales, en los que la pleura aparece engrosada y los pulmones acartonados y con pequeñas cavidades quísticas llenas de aire. Las pequeñas y grandes vías aéreas suelen estar deformadas, y son frecuentes las bronquiolectasias y bronquiectasias, respectivamente7. Por lo tanto, al principio la NI en la ES (NI-ES) se consideraba un simple proceso fibrosante indistinguible de la fibrosis pulmonar idiopática (FPI). Antes de la clasificación de Katzenstein y Myers, se pensaba que sus histologías eran idénticas, aunque se sabía que la NI-ES se asociaba a un mejor pronóstico que la forma idiopática8.
Se han reclasificado las neumopatías intersticiales idiopáticas9 identificando el diagnóstico histopatológico de neumonía intersticial usual (NIU) con el diagnóstico clínico de FPI. Aunque la NIU se puede observar en pacientes con NI-ES, el patrón histopatológico que más frecuentemente se encuentra en los pacientes con ES es la neumonía intersticial no específica (NINE). La distinción entre NIU y NINE tiene gran importancia terapéutica y pronóstica pero, desgraciadamente, esta diferenciación puede resultar difícil. De hecho, no se sabe si la NINE representa un estadio inicial de la NIU, ya que se puede encontrar ambos patrones en biopsias de un mismo paciente, e incluso en múltiples biopsias de un mismo lóbulo. Además, hay una variabilidad interobservador significativa, incluso entre patólogos expertos en el diagnóstico de estas entidades4.
En un estudio10 de tejido pulmonar de autopsias de pacientes con ES, se encontraron dos alteraciones celulares importantes en los estadios iniciales de NI-ES: una fue la aparición de un gran número de miofibroblastos que expresaban alfaactina de músculo liso en el intersticio y la otra la excesiva formación de capilares alveolares irregulares (neovascularización), junto con un aumento del número de células endoteliales microvasculares. De todas formas, a medida que progresa la fibrosis hacia su estadio final, disminuye la población de miofibroblastos y células endoteliales capilares. Estas alteraciones temporales indican que la proliferación miofibroblástica y las alteraciones vasculares pueden ser relevantes en la etiopatogenia de la NI-ES.
PATOGENIA
La patogenia de la NI-ES es compleja e implica alteraciones en los fibroblastos, células endoteliales y células del sistema inmunitario. Estas alteraciones frecuentemente conducen a una fibrosis grave y progresiva, enfermedad macrovascular y microvascular, así como a la activación de la inmunidad humoral y celular, con la producción de autoanticuerpos, citocinas, quimiocinas y factores de crecimiento2.
Autoanticuerpos
Los anticuerpos antinucleares se detectan en más del 90% de los pacientes con ES. Los valores de anticuerpos antitopoisomerasa I se correlacionan con la actividad y la gravedad de la enfermedad y se asocian a la NI-ES. También se ha relacionado con la afección pulmonar los anticuerpos antifibroblasto, antimetaloproteinasas de la matriz (IgG anti-MMP-3), y el FR-IgA2,4,11.
Citocinas y quimiocinas
Los macrófagos alveolares son el tipo celular más frecuente en el infiltrado inflamatorio de la NI-ES activa. Parece que los macrófagos en la NI-ES se activan por una vía alternativa, en vez de por la clásica. Habitualmente los macrófagos son activados por lipopolisacáridos, el factor de necrosis tumoral alfa (TNF*), el interferón gamma (IFN*), la interleucina (IL) 12 o la ingestión de material necrótico, que hacen que éstos produzcan grandes cantidades de citocinas proinflamatorias, como TNF*, IL-1 e IL-6, pero poca cantidad de factor de crecimiento tumoral beta (TGFß). Estos macrófagos son eficientes como presentadores de antígeno, preferentemente activan los linfocitos Th1 e inhiben la producción de colágeno por los fibroblastos. Sin embargo, en la ES los macrófagos son activados por IL-4, TGFß, glucocorticoides (GC) o fagocitosis de material apoptótico; tienen un fenotipo antiinflamatorio, fagocítico y profibrótico y activan fundamentalmente linfocitos Th2. Estos macrófagos activados por una vía alternativa producen grandes cantidades de TGFß, del factor de crecimiento derivado de las plaquetas (PDGF) y del antagonista del receptor de IL-1 (IL-1ra), pero no de TNF* o IL-1. También producen quimiocinas, como el factor quimiotáctico monocitario (MCP) 1 y la quimiocina pulmonar regulada por la actividad (PARC). Los macrófagos activados por la vía alternativa estimulan la formación de colágeno por los fibroblastos. Por tanto, estos macrófagos parecen ser un punto clave en la patogenia de la NI-ES, ya que son activados por factores profibróticos (TGFß, IL-4), producen factores profibróticos (TGF*, MCP-1 y PARC) y activan linfocitos Th2 profibróticos productores de IL-4.
En la NI-ES también está aumentada la producción del factor de crecimiento del tejido conectivo (CTGF), que es un mediador de los efectos del TFGß y de la oncostatina M (OSM).
El aumento de mastocitos se asocia a valores elevados de histamina en el lavado broncoalveolar (LBA) de pacientes con NI-ES. Los eosinófilos se adhieren a los fibroblastos y directamente los activan y estimulan la producción de colágeno a través de la TFGß11.
Daño microvascular
La trombina, la endotelina (ET) 1, la tromboglobulina ß (BTG) y el factor plaquetario 4 (PF4) indican daño microvascular en la NI-ES. La trombina diferencia los fibroblastos normales del pulmón hacia miofibroblastos. La ET-1 es un mediador de fibrosis y está elevada en el plasma y en los fibroblastos dérmicos de pacientes con ES. La BTG y el PF4 son proteínas específicas liberadas por las plaquetas activadas, y por tanto se consideran marcadores de activación plaquetaria. Las plaquetas participan en la inflamación a través de la liberación de factores quimiotácticos y producción de eicosanoides proinflamatorios. Las concentraciones de BTG y PF4 se detectan principalmente en las primeras fases de la NI-ES2,10.
Moléculas de adhesión
Las formas solubles de algunas moléculas de adhesión, que regulan el reclutamiento linfocitario, están elevadas en sueros de pacientes con ES. Sin embargo, no todas ellas se relacionan con la NI-ES. Por ejemplo, se ha relacionado la glucoproteína ligando de la selectina P (sPSGL-1) con un menor riesgo de NI-ES, ya que sus concentraciones se correlacionan de forma directa con la CVF y la DLCO2.
MANIFESTACIONES CLINICAS
Aunque la afección pulmonar suele ser precoz en la ES, un elevado porcentaje de pacientes permanecen asintomáticos en las primeras etapas de la enfermedad. Los síntomas suelen aparecer más tardíamente y consisten en disnea, astenia y tos seca. La disnea normalmente es leve, y en relación con el esfuerzo. Su intensidad no se correlaciona con el grado de fibrosis en la radiografía de tórax. La tos seca suele aparecer aproximadamente en el 15% de los pacientes y también puede estar causada por el reflujo gastroesofágico (RGE), bronquitis, bronquiectasias y neumonía por aspiración. En algunos casos puede aparecer sibilancias o dolor pleurítico. La hemoptisis es rara y, si aparece, obliga a descartar neoplasia o sangrado por telangiectasias. También hay que descartar las infecciones y la toxicidad por fármacos.
El hallazgo más característico en la auscultación pulmonar son los crepitantes secos basales. Se ha propuesto que la reducción del flujo sanguíneo digital explicaría la baja frecuencia de acropaquias.
También pueden contribuir al deterioro de la función respiratoria la esclerosis cutánea grave del tronco y la miositis de los músculos respiratorios, al condicionar hipoventilación. Con el progresivo deterioro del parénquima pulmonar, acaba apareciendo hipertensión pulmonar secundaria y cor pulmonale3,7.
PRUEBAS COMPLEMENTARIAS
El tratamiento de la NI-ES sólo es útil en estadios precoces, en los que se puede estabilizar la enfermedad. Por ello es necesario un diagnóstico temprano y una valoración de la relevancia clínica de la afección pulmonar, que incluya la gravedad y las posibilidades de progresión. Las decisiones terapéuticas se apoyarán en esta información12. El diagnóstico se basa fundamentalmente en los resultados de las pruebas de función respiratoria, la TACAR de tórax y el LBA. En la tabla 1 se recoge una propuesta de elección de pruebas complementarias para los pacientes con ES, atendiendo a diferentes escenarios clínicos.

Radiografía de tórax
Aunque la radiografía de tórax es poco sensible para la detección de la NI-ES, se debe realizar al inicio, con el objeto de excluir otras afecciones, como neoplasias, infecciones o neumonía por aspiración3. Normalmente aparece alterada en un 25-61% de los pacientes con NI-ES, y suele mostrar cambios reticulonodulares bibasales indistinguibles de la FPI. La afección pleural es rara, por lo que si aparece, obliga a descartar otras causas. El grado de afección radiológica no se correlaciona con la afección clínica ni con los valores de las PFR7.
Pruebas de función respiratoria
Son útiles para detectar y seguir la evolución de la enfermedad. Aparecen alteradas entre un 45 y un 100% de los pacientes con ES, según las series4. Se debe tener en cuenta que otros factores, como el tabaco, la miositis, la fibrosis de la pared torácica o la coexistencia de hipertensión pulmonar también pueden alterar sus resultados7. Se ha comprobado que los valores de las PFR que mejor se correlacionan con el grado de afección pulmonar son la capacidad de difusión de monóxido de carbono (DLCO) y la CVF. El hallazgo más común es un trastorno ventilatorio restrictivo con disminución de la DLCO, que aparece en el 30-50% de los pacientes con síntomas respiratorios. La alteración inicial suele ser la disminución de la DLCO13, que suele ir ya asociada a un patrón restrictivo cuando alcanza valores por debajo del 65% de la esperada. En este contexto, unos volúmenes pulmonares normales indican hipertensión pulmonar.
Se pueden encontrar PFR alteradas en pacientes asintomáticos y con radiografía de tórax e incluso TACAR normales. Por ello es importante realizar unas PFR basales y seriadas durante los primeros 5 años de la enfermedad, ya que la clínica y la radiografía de tórax son poco sensibles para detectar la afección pulmonar. El empeoramiento más rápido se produce en los primeros 2 años de la enfermedad y se ha demostrado que una DLCO < 40% condiciona un mal pronóstico5. La desaturación con el ejercicio suele ocurrir en etapas muy tempranas. Es recomendable realizar un test de la marcha cuando se diagnostica la ES, además de la espirometría y la DLCO4.
Gammagrafía con DTPA-99mTc
El aclaramiento de dietilentriaminpentaacetato (DTPA) marcado con 99mTc del pulmón es una medida de la permeabilidad epitelial, que está incrementada en la inflamación, así como de la fibrosis de la barrera epitelial pulmonar. Se ha visto que en la NI-ES, el aclaramiento de DTPA-99mTc está incrementado antes de que la enfermedad sea detectada radiológicamente, y este incremento es un marcador de progresión durante los años siguientes14.
Tomografía computarizada de alta resolución torácica
La TACAR detecta la NI con una sensibilidad entre el 90 y el 100%. Actualmente es el estándar de la técnica no invasiva para el diagnóstico de NI. El tipo de afección en la TACAR puede predecir el patrón histológico, la progresión de la enfermedad y la respuesta al tratamiento.
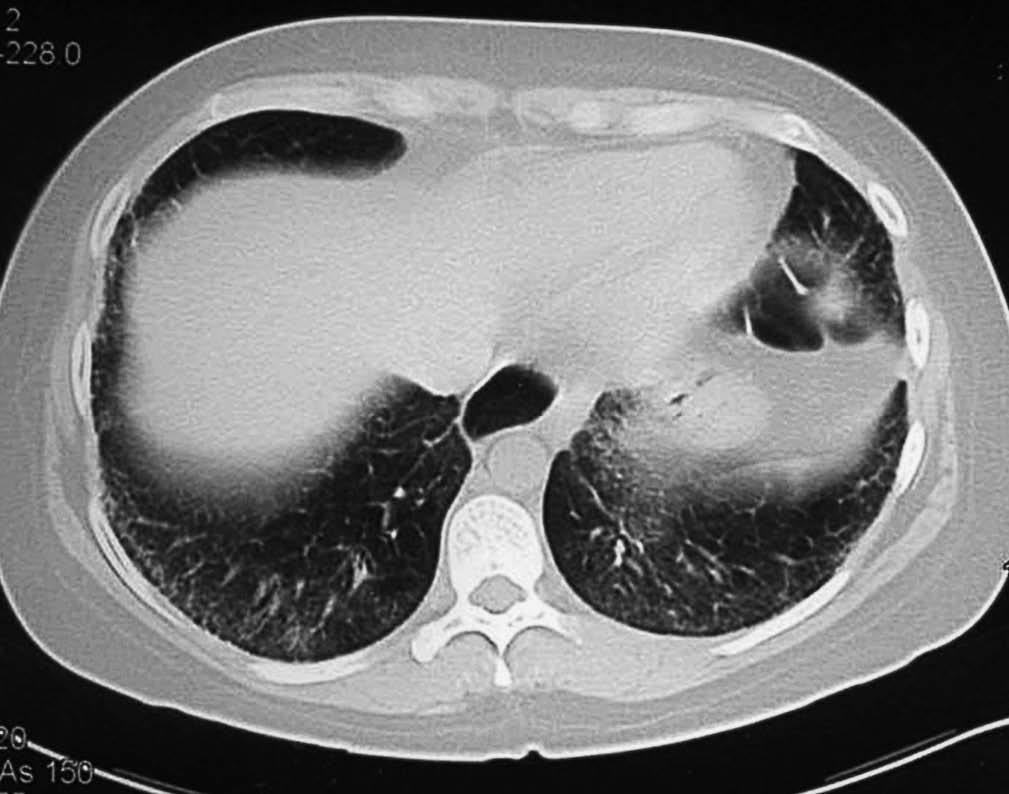
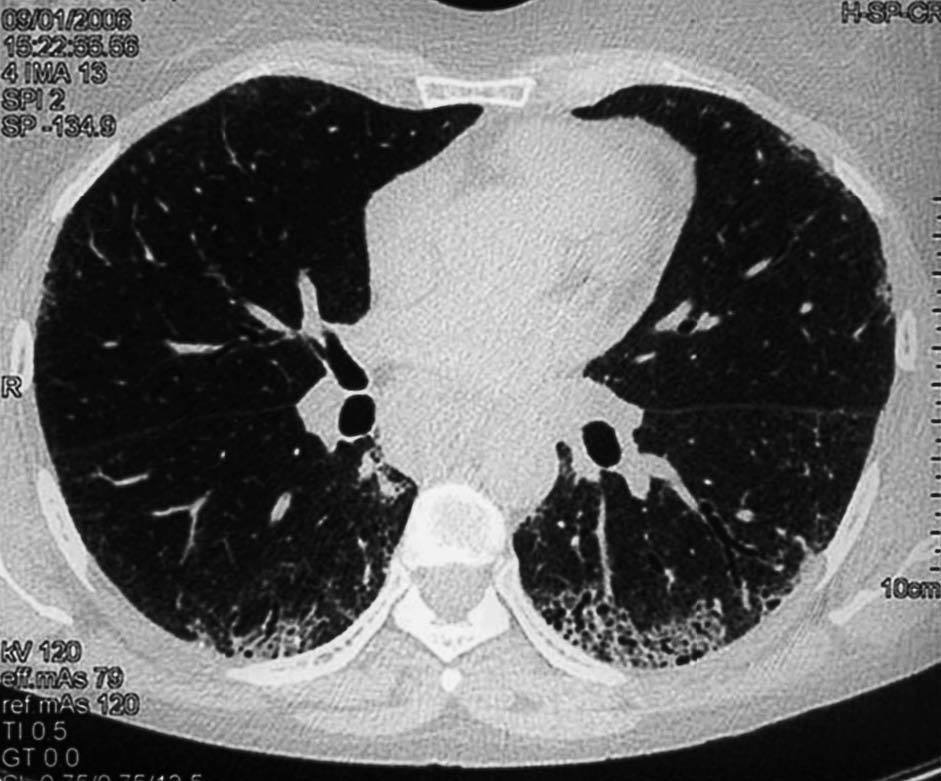
Los hallazgos principales de afección parenquimatosa son el vidrio esmerilado, las opacidades lineales irregulares, el panal de abeja y las bronquiectasias de tracción. Además se puede encontrar micronódulos subpleurales, dilatación esofágica, elongación de las arterias pulmonares, afección pleural y adenopatías mediastínicas15. El patrón en vidrio esmerilado (fig. 1) aparece como un aumento de opacidades borrosas en el parénquima pulmonar, con preservación de la señal bronquial y vascular. El patrón en panal de abeja (fig. 2) se caracteriza por innumerables sombras lineales entrelazadas, que pueden ser finas o más gruesas, con importante desestructuración de la arquitectura pulmonar16.
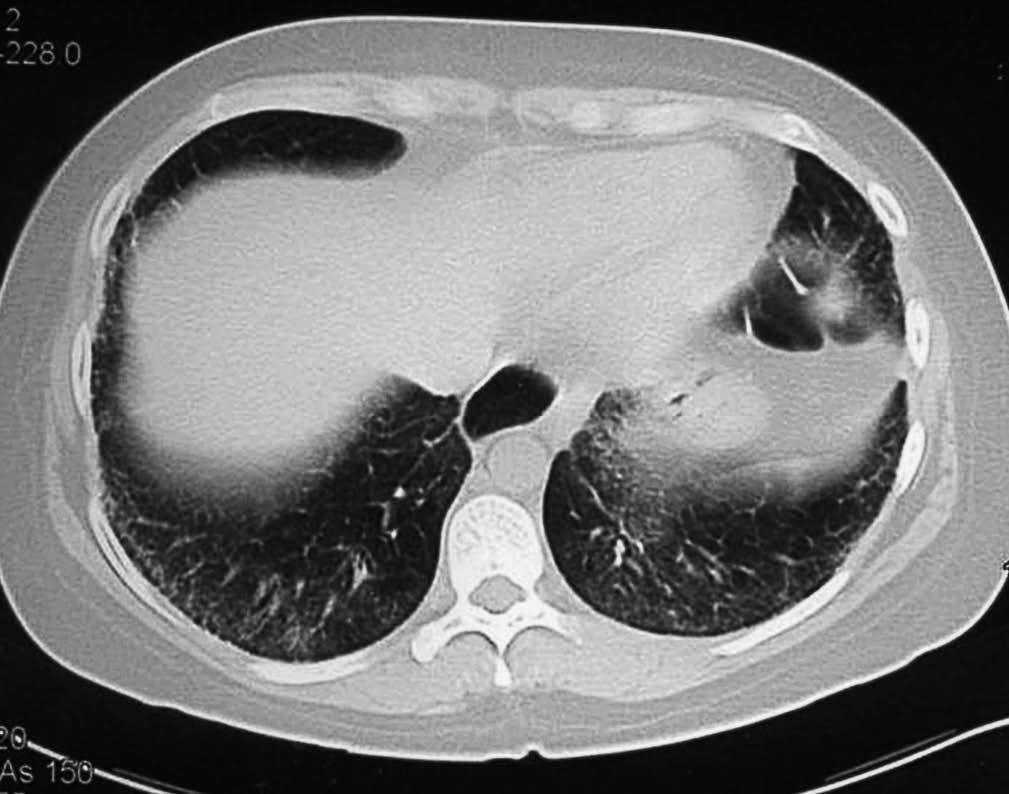
Figura 1 Tomografía axial computarizada de alta resolución de una mujer de 34 años con esclerosis sistémica y disnea de moderados esfuerzos de reciente aparición. Se observan zonas de vidrio esmerilado, indicio de alveolitis activa, de predominio subpleural y basal.
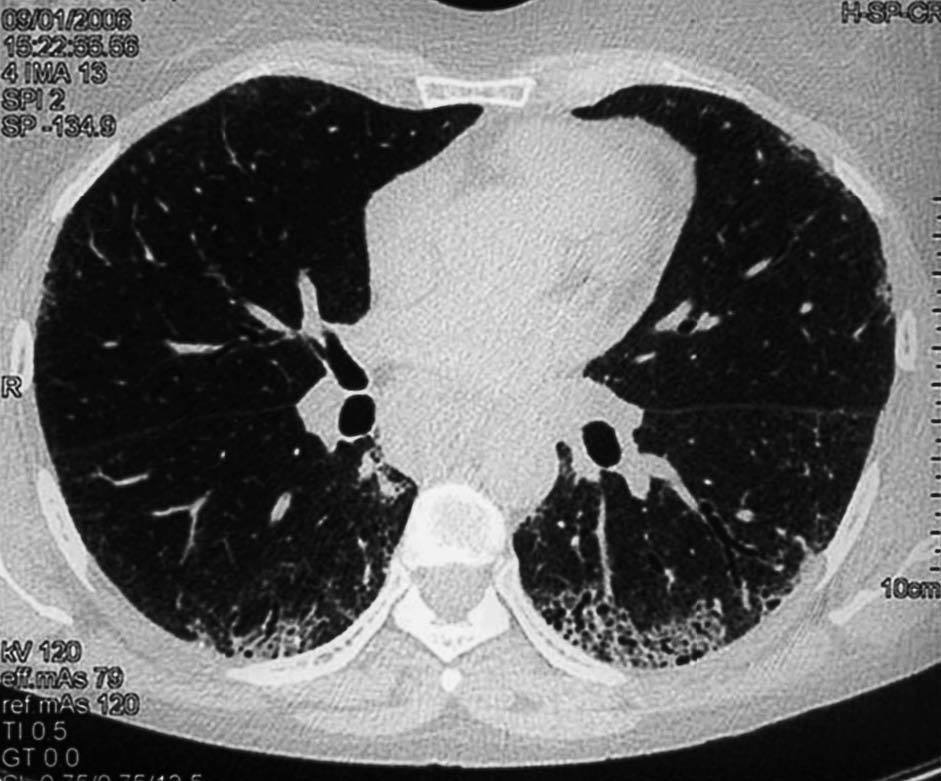
Figura 2 Tomografía axial computarizada de alta resolución de una mujer de 43 años con esclerosis sistémica y disnea de mínimos esfuerzos. Se observan zonas de panalización, típicas de fibrosis pulmonar avanzada.
El diagnóstico por TACAR hace innecesaria, en la mayoría de los casos, la biopsia pulmonar. Aun así, en un estudio17 en el que se compararon los hallazgos en la TACAR de pacientes con diagnóstico histopatológico de NIU y de NINE, se vio que había una superposición considerable de los patrones de ambas en la TACAR, que conducía a un diagnóstico erróneo de NINE en el 30% de los casos. Este estudio demostró que la NINE se asocia a un patrón reticular más fino y a una mayor proporción de vidrio esmerilado en la TACAR que la NIU, pero que no hay diferencias en cuanto a la distribución de la afección pulmonar radiológica entre ambos patrones histológicos.
En otro estudio16 publicado por el mismo grupo, se compararon los hallazgos en la TACAR de pacientes con NI-ES, con FPI y con NINE. Se vio que el patrón de afección pulmonar en la TACAR en pacientes con NI-ES era distinto del de los pacientes con FPI, ya que los primeros presentaban una fibrosis menos tosca y una mayor proporción de vidrio esmerilado. Sin embargo, el patrón de afección en la NI-ES era muy parecido al de los pacientes con NINE.
Se ha intentado correlacionar evolutivamente los cambios morfológicos en la TACAR con los cambios en las PFR. En una serie15 de 40 pacientes con NI-ES se observó que la única correlación significativa se daba entre el aumento del patrón en panal de abeja y el descenso de la DLCO. En otra serie previa18 de 39 pacientes con FPI, se pudo correlacionar los cambios en la TACAR no sólo con la DLCO, sino también con la CVF.
Lavado broncoalveolar
En casos seleccionados, el LBA también puede ser útil en la aproximación diagnóstica a la NI-ES. Las alteraciones en el LBA suelen preceder a los cambios en la TACAR19. Sin embargo, el papel del LBA en el diagnóstico de NI-ES no está del todo claro, en parte debido a la falta de un método estandarizado para la recogida y análisis del material obtenido en el lavado20.
Se considera que la NI-ES es un proceso de alveolitis que evoluciona hacia la fibrosis pulmonar. Uno de los cambios más tempranos, que indica alveolitis en pacientes con ES, es el elevado recuento de células inflamatorias en el LBA5. En diversos estudios se ha asociado el recuento elevado de granulocitos en el LBA con enfermedad progresiva, sobre todo en los pacientes no tratados21-23. Los pacientes con un recuento elevado de neutrófilos o de eosinófilos en el LBA presentan una reducción significativa de la DLCO y de la CVF, mayor grado de disnea y alteraciones radiológicas más marcadas a los 2 años de seguimiento que los pacientes que presentan un predominio de linfocitos o un LBA normal21,23-26. Se ha sugerido que la neutrofilia en el LBA se relaciona más con enfermedad extensa, mientras que la eosinofilia lo hace con enfermedad progresiva, independientemente de su extensión12. Parece razonable pensar que la granulocitosis en el LBA indica un estado activo de la NI-ES, mientras que la linfocitosis o el LBA normal aparecerían en fases inactivas de la enfermedad. Esto significaría que la NI-ES no es necesariamente una enfermedad uniformemente progresiva, sino que tiene un curso ondulante, con brotes y remisiones, al igual que ocurre en otras enfermedades reumáticas autoinmunitarias. En relación con estos hallazgos, Witt et al25 señalan que debería valorarse iniciar tratamiento inmunosupresor en los pacientes con granulocitosis en el LBA, y adoptar una actitud expectante cuando el recuento celular mayoritario sea de linfocitos.
Clements et al27 publicaron un estudio en el que comparaban los hallazgos del LBA y la TACAR en distintos lóbulos pulmonares (língula y segmentos inferiores) en 15 pacientes con NI-ES. Encontraron una alta concordancia entre el vidrio esmerilado de la TACAR y la alveolitis en el LBA en la língula, pero no en los lóbulos inferiores. La correlación entre la fibrosis en la TACAR y la alveolitis en el LBA fue significativa para los lóbulos inferiores, pero no para los lóbulos medios. Los autores recomiendan realizar el LBA en al menos 2 segmentos pulmonares, además de la TACAR, para el diagnóstico de alveolitis. Según los resultados de otro estudio4, la TACAR es más sensible que el LBA para la detección de NI-ES (el 90,6 contra el 70%) y la concordancia entre la TACAR y el LBA es sólo del 65%. Por tanto, no se debería tomar decisiones terapéuticas basadas únicamente en los hallazgos del LBA, aunque éste, ocasionalmente, sí puede tener un papel importante en la exclusión de otros procesos, en particular las infecciones y las neoplasias3,27.
También se ha estudiado28 el perfil de las citocinas en el LBA de pacientes con NI-ES, comparándolo con el de pacientes con NIU. Los pacientes con NI-ES presentan un balance más favorable entre las citocinas fibróticas (MCP-1) y las antifibróticas (IL-10 e IL-12) que los pacientes con NIU, lo que podría explicar el mejor pronóstico de los primeros. En otro estudio29 se demostró que en la NI-ES hay una sobreproducción de leucotrienos (LTB4), proinflamatorios y profibróticos, que no está compensada por un incremento en la producción de lipoxinas antiinflamatorias y antifibróticas, como 15-HETE o el LXA4.
Biopsia pulmonar
Rara vez es necesaria la realización de biopsia pulmonar por toracoscopia para el diagnóstico de NI-ES. Sólo se recomienda en los casos con imágenes atípicas en la TACAR o para descartar enfermedad granulomatosa. Dado que la enfermedad tiene una distribución predominantemente subpleural, es preferible realizar una biopsia pulmonar periférica. La biopsia transbronquial tiene escasa rentabilidad, ya que la naturaleza parcheada de la enfermedad hace que la muestra de tejido obtenido sea insuficiente para el diagnóstico adecuado de fibrosis. Esto puede ocurrir incluso con la biopsia mediante toracoscopia. Actualmente, con la TACAR, la evaluación histopatológica añade muy poco al diagnóstico y a la valoración pronóstica de la NI-ES3,12,30,31.
Otros marcadores de enfermedad pulmonar
Como se ha comentado anteriormente, se relacionan con NI-ES los anticuerpos antitopoisomerasa I, antifibroblasto, antimetaloproteinasas de la matriz (IgG anti-MMP-3) y el FR-IgA2,4,11.
La ES se caracteriza por importantes alteraciones microvasculares. La capilaroscopia permite evaluar de manera no invasiva la disfunción vascular. Se ha sugerido que las alteraciones en la capilaroscopia en pacientes con ES de menos de 5 años de evolución se relacionan con la presencia de opacidades en vidrio esmerilado en el pulmón. Esto convertiría a la capilaroscopia en una técnica útil para la detección precoz de pacientes con riesgo de desarrollar NI-ES32.
En la NI-ES hay una marcada hiperplasia de los neumocitos tipo II, que producen y secretan las proteínas del surfactante. Se ha observado que las proteínas A y D están elevadas en la NI-ES y que sus valores se correlacionan con la actividad de la enfermedad33.
La KL-6 es una glucoproteína producida fundamentalmente por los neumocitos tipo II, pero también por los macrófagos alveolares. Sus valores se encuentran elevados en los pacientes con NI-ES34.
El óxido nítrico exhalado, probablemente producido por células inflamatorias como los macrófagos alveolares, está aumentado en los pacientes con NI-ES y reducido en los pacientes con hipertensión pulmonar. Dado que se ha encontrado elevado en la NI-ES subclínica detectada por LBA, este marcador no invasivo podría usarse para la detección precoz de la NI-ES4.
TRATAMIENTO
Las recomendaciones para el tratamiento de la NI-ES se han basado, fundamentalmente, en estudios retrospectivos y no controlados y en protocolos terapéuticos extrapolados de la FPI. Clásicamente, los corticoides suponían la base fundamental del tratamiento, al igual que en otras formas de fibrosis pulmonar, a pesar de la falta de evidencia de su eficacia y la preocupación por el riesgo de precipitar crisis renales. Esta preocupación procedía, en parte, de un estudio caso-control35 en el que el uso de al menos 15 mg/día de prednisona durante 6 meses se asociaba a una mayor prevalencia de crisis renales esclerodérmicas. En un estudio más reciente36, en el que se incluyó a 134 pacientes con ES difusa inicial, se observó que la terapia con prednisona se asociaba a la crisis renal esclerodérmica independientemente de otros factores de riesgo, como la afección cutánea grave, las contracturas de grandes articulaciones o la cardiomegalia en la radiografía de tórax. Pero esta asociación sólo ocurría en los pacientes que tenían simultáneamente afección cutánea grave y contracturas de grandes articulaciones. En el estudio de Pakas et al37 se compararon las dosis altas de prednisolona (1 mg/kg/día durante 4 semanas, seguido de una pauta descendente) con dosis bajas (< 10 mg/día), añadidas al tratamiento con pulsos intravenosos de ciclofosfamida (CF), en 28 pacientes con NI-ES. A los 6 meses de seguimiento, se observó mejoría de la CVF y de la DLCO en el grupo de dosis altas, que se mantenía a los 12 meses, mientras que en el grupo de dosis bajas no mejoró ninguno de los criterios de valoración. Este estudio presenta algunas limitaciones, ya que la asignación del tratamiento no fue aleatoria y los pacientes del grupo de dosis bajas tenían una enfermedad más grave y evolucionada. Todos los pacientes del grupo de dosis altas desarrollaron hábito cushingoide durante los primeros meses de tratamiento, pero en ninguno se desarrolló una crisis renal esclerodérmica durante el estudio. Tampoco hubo retiradas por efectos adversos. En definitiva, no está claro si la terapia corticoidea contribuye a precipitar las crisis renales en los pacientes con ES o es simplemente un marcador de enfermedad más grave.
La D-penicilamina ha sido uno de los fármacos más utilizados en el tratamiento de la ES. Datos procedentes de estudios retrospectivos38,39 indicaban que podía ser beneficiosa en la NI-ES, al mejorar la DLCO e incluso la CVF. En un estudio prospectivo más reciente, Clements et al40 compararon dosis altas de D-penicilamina (750-1000 mg/día) con dosis bajas (125 mg/48 h) en 68 pacientes con ES de forma difusa de menos de 18 meses de evolución. Los autores no encontraron diferencias en la gravedad y la extensión de la afección cutánea ni en la aparición de complicaciones renales o pulmonares entre ambos grupos. El tratamiento se retiró en casi el 50% de los pacientes debido a los efectos secundarios. Al cabo de 2 años de seguimiento, no hubo mejoría de la DLCO ni de la CVF en ninguno de los grupos. Posteriormente, se ha publicado otro estudio retrospectivo41 que tampoco ha conseguido demostrar un efecto beneficioso de la D-penicilamina en el tratamiento de la NI-ES.
La azatioprina ha demostrado su eficacia y seguridad en la FPI42, pero los datos publicados en la NI-ES son escasos. En el estudio retrospectivo de Steen et al43, en el que se comparaban diferentes inmunosupresores en 122 pacientes con NI-ES, los tratados con azatioprina y metotrexato presentaron los peores resultados, aunque parece que al inicio tenían una enfermedad más grave. En un reciente estudio retrospectivo44 se incluyó a 11 pacientes con NI-ES que habían recibido tratamiento con azatioprina y dosis bajas de GC; 3 pacientes interrumpieron el tratamiento antes de los 6 meses por efectos secundarios. En lte significativa en los índices de disnea. La CVF mejoró en 5 de ellos y se mantuvo estable en 3, pero estos resultados no alcanzaron significación estadística. Lo importante es que no hubo deterioro de los síntomas ni de la CVF en los pacientes que completaron los 12 meses de tratamiento. Además, la azatioprina fue efectiva en 3 pacientes que habían recaído tras recibir tratamiento con CF. Hasta la fecha no hay otros estudios que evalúen la eficacia de la azatioprina en la NI-ES, pero se ha usado ampliamente en la FPI, con buena tolerancia.
El clorambucilo se ha utilizado sin éxito en la ES, tanto para las manifestaciones cutáneas como para las viscerales45. El metotrexato también se ha utilizado en el tratamiento de la ES y, aunque pudiera ser efectivo para mejorar la afección cutánea46, no ha resultado beneficioso en la NI-ES43. El uso de la ciclosporina A, que podría mejorar las manifestaciones cutáneas de la ES, se ha visto limitado por su potencial nefrotoxicidad46.
Otro inmunosupresor utilizado en el tratamiento de la NI-ES es el micofenolato mofetilo (MMF). Liossis et al47 han publicado un estudio abierto prospectivo con 6 casos de NI-ES, en los que usaron MMF por su menor toxicidad frente a la CF. De los 6 pacientes, 5 presentaban una NI-ES precoz y el otro era un caso avanzado resistente a CF. Todos recibieron MMF, con incrementos secuenciales de la dosis hasta alcanzar los 2.000 mg/día y 7,5-10 mg/día de prednisolona. A los 6 meses, la DLCO había mejorado significativamente en todos ellos y la CVF en todos, salvo en un paciente con enfermedad de reciente comienzo. Los síntomas respiratorios mejoraron a partir del tercer mes en los 5 pacientes con enfermedad precoz y el patrón en vidrio esmerilado en la TACAR mejoró en 4 de los 5 pacientes con enfermedad de reciente comienzo. No hubo casos de infección relevante ni de leucocitopenia. Los autores consideran que el MMF representa una alternativa al tratamiento con CF, ya que no sólo es un fármaco seguro y bien tolerado, sino que en su serie fue eficaz en mejorar la DLCO, la TACAR, los síntomas respiratorios y la calidad de vida de los pacientes.
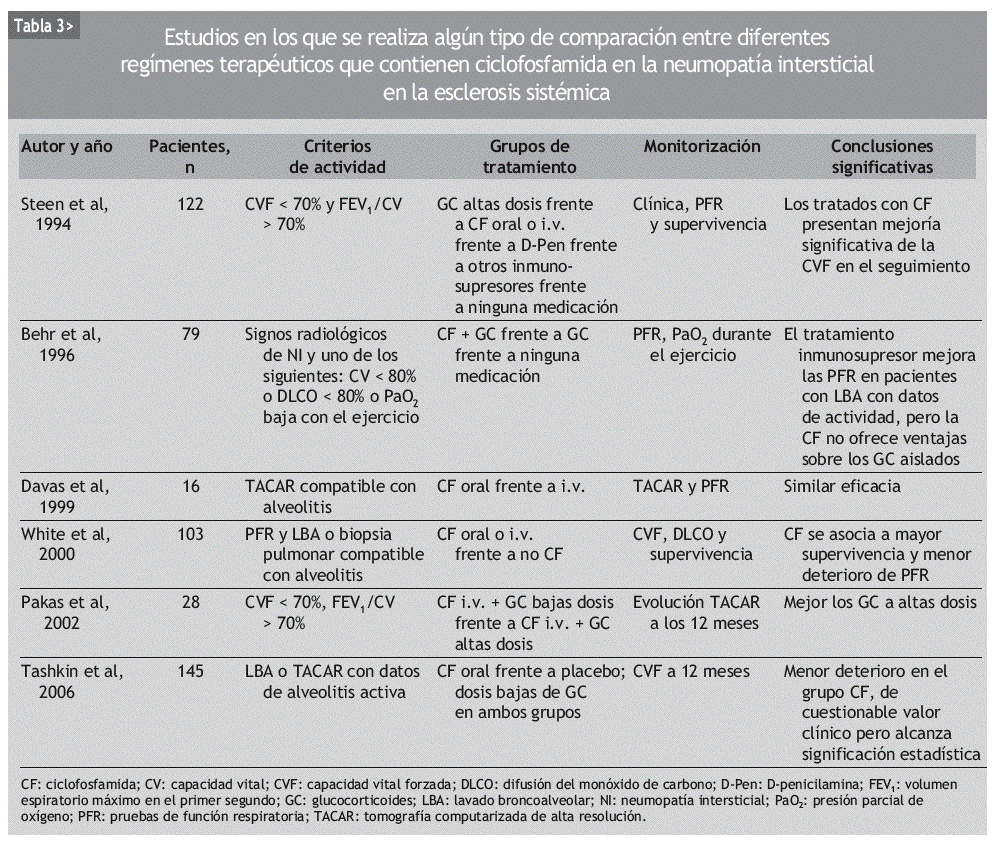
Aunque se ha usado empíricamente diversos inmunosupresores en el tratamiento de la NI-ES, la mayor parte de los datos publicados se refiere a la CF. Se trata de numerosas series de casos y comparaciones históricas, en las que se ha observado mejoría de la NI-ES tratada con CF, según diferentes parámetros clínicos, radiológicos, espirométricos y de DLCO. La mayoría incluye grupos de pacientes heterogéneos, no controlados por la gravedad o la duración de la enfermedad (tablas 2 y 3).
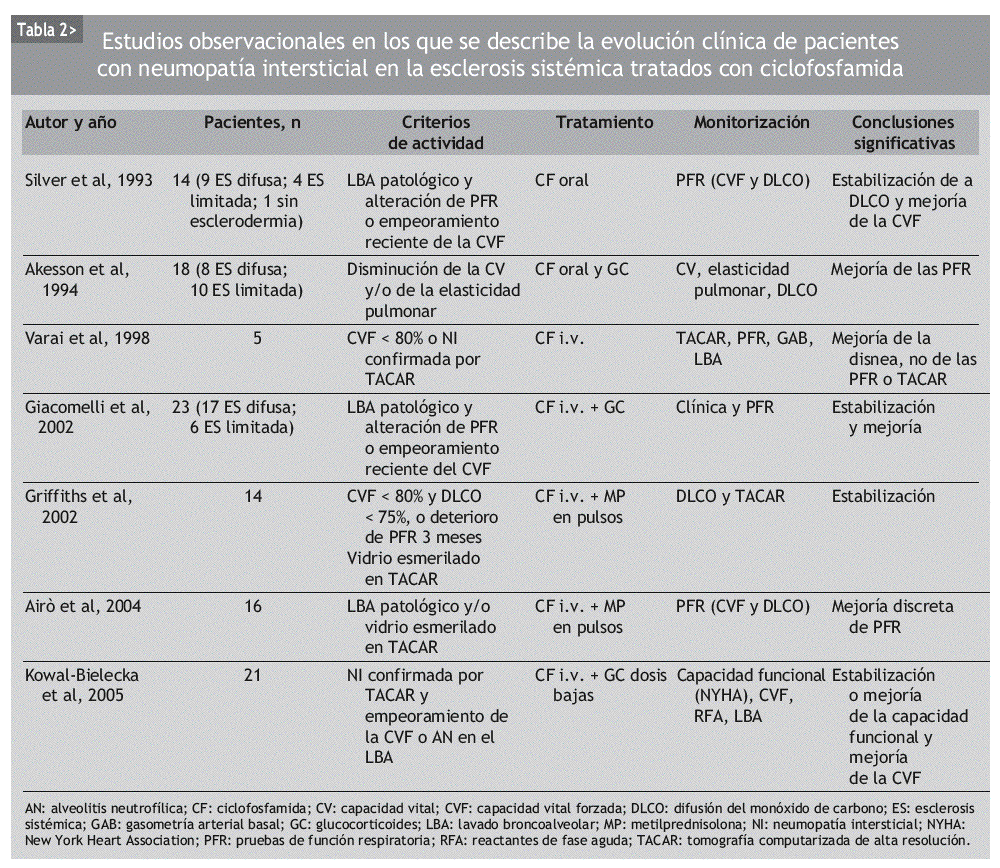
En 1993, Silver et al48 publicaron los resultados de un estudio prospectivo no controlado, en el que incluyeron a 14 pacientes con NI-ES, atendiendo a los hallazgos del LBA junto con un empeoramiento de las PFR. Fueron tratados con CF oral diaria y dosis bajas de GC durante un máximo de 2 años. A los 6 meses de seguimiento, la CVF mejoró en 6 de ellos, se mantuvo estable en 7 y empeoró en 1. A los 12 meses, la mayoría mantenía la mejoría alcanzada. La DLCO no mejoró sustancialmente durante el seguimiento, pero permaneció estable en la mayoría de los casos. Cinco pacientes completaron los 2 años de tratamiento. Hubo una retirada por neumonía a los 12 meses y 2 por cistitis hemorrágica, a los 6 y a los 12 meses.
Un año más tarde, Akesson et al49 publicaron un estudio que incluía a 18 pacientes con NI-ES, atendiendo a las PFR alteradas y/o evidencia radiológica de afección pulmonar. Los pacientes fueron asignados a dos grupos distintos, basándose en la presencia o ausencia de reactantes de fase aguda elevados, y tratados con una dosis inicial diaria de CF oral de 2-2,5 mg/kg y 30 mg de prednisolona, que fue reduciéndose en un período de 10 semanas, hasta alcanzar los 5-10 mg/día. La dosis de CF tuvo que ser reducida en 16 de los 18 pacientes por leucocitopenia y trombocitopenia, y retirada en un paciente por hematuria macroscópica. Tras un año de tratamiento, la CVF había mejorado en 14 pacientes (p = 0,01) y la elasticidad pulmonar en 8 de los 12 en los que se había determinado (p = 0,06). El análisis por subgrupos puso de manifiesto que esta mejoría era más prominente en los pacientes que inicialmente tenían reactantes de fase aguda elevados. La afección cutánea también había mejorado al final del tratamiento.
En 1994, Steen et al43 comunicaron los resultados de un estudio observacional retrospectivo con 122 pacientes en el que se comparaban altas dosis de prednisona sola, CF, D-penicilamina, otros inmunosupresores (azatioprina o metotrexato) y no tratar. En los últimos 4 grupos, algunos pacientes fueron tratados con GC. Los pacientes incluidos presentaban un trastorno ventilatorio restrictivo moderado, definido como una CVF < 70% de la esperada y un cociente FEV1/CVF > 70%. De los 5 grupos, el que recibió CF experimentó una mejoría en la CVF significativamente mayor que los demás grupos. El grupo de otros inmunosupresores fue el que obtuvo los peores resultados. El único factor predictor de mejoría significativa, independientemente del fármaco utilizado, resultó ser una duración de la enfermedad inferior a los 4 años en el momento de iniciar el tratamiento.
En 1996, Behr et al22 publicaron un estudio prospectivo de 79 pacientes con deterioro de la función pulmonar en las PFR. De ellos, 41 no se trataron, 24 recibieron GC y 14 pacientes recibieron GC y CF. Los autores concluyeron que el tratamiento inmunosupresor mejoraba las PFR, sobre todo en los pacientes con datos de actividad en el LBA. Por otra parte, añadir CF a los pacientes que no habían respondido al GC aportaba un beneficio muy discreto.
En 1998, Varai et al50 publicaron los datos de 5 pacientes con NI-ES confirmada por PFR o TACAR, que recibieron tratamiento con 1 g de CF por vía intravenosa (i.v.) mensual durante 48 semanas. A diferencia de otros estudios, ningún paciente recibió GC. Al final del estudio, todos los pacientes habían experimentado una mejoría clínica de la disnea, pero la CVF, la DLCO y los hallazgos de la TACAR permanecieron estables. Todos los pacientes desarrollaron leucocitopenia y hubo un caso de cistitis hemorrágica, pero no fue necesario retirar el fármaco a ningún paciente.
En 1999, Davas et al51 publicaron los resultados de un estudio en el que comparaban la eficacia de la CF oral y de la i.v. en el tratamiento de 16 pacientes con TACAR compatible con alveolitis. Ambos grupos recibieron 10 mg/día de prednisona. Tras revaluar a los pacientes a los 6 y 12 meses, los autores concluyeron que tanto la CF oral como la i.v. mejoraban a los pacientes en un estadio precoz de la enfermedad, identificable por un patrón en vidrio esmerilado en la TACAR. Pero los cambios en la extensión de la enfermedad por TACAR tras 12 meses se correlacionaban con la DLCO sólo en el grupo de la CF i.v.
White et al23 publicaron en el año 2000 un análisis retrospectivo de 103 pacientes con ES en los que se había realizado PFR y LBA o biopsia pulmonar. De los 69 sujetos que tenían alveolitis, 39 habían recibido terapia con CF. Comparando los 3 grupos de pacientes (sin alveolitis, con alveolitis tratados, con alveolitis no tratados), los autores señalan que el tratamiento con CF estabiliza la función pulmonar y mejora la supervivencia en los pacientes con alveolitis respecto a los no tratados.
Pakas et al37 publicaron en 2002 el estudio descrito con anterioridad en el que comparaban dosis altas y dosis bajas de GC añadidas a la CF en el tratamiento de 28 pacientes con NI-ES.
Giacomelli et al52 presentaron el mismo año una serie de 23 pacientes con PFR y LBA alterados, que habían recibido 6 ciclos mensuales de 1 g/m2 de CF y 25 mg diarios de prednisona oral durante un mes, con posterior pauta descendente. En la valoración a los 6 meses, la CVF había permanecido estable y la DLCO había mejorado sin alcanzar significación estadística. La disminución en el recuento celular del LBA y la mejoría en la TACAR tampoco resultaron estadísticamente significativas. Tras el tratamiento, no se encontró correlación entre la afección por TACAR y el LBA. Los autores concluyen que el tratamiento combinado con CF i.v. y dosis bajas de GC estabiliza e incluso puede mejorar la función pulmonar en los pacientes con NI-ES, mientras que los hallazgos de la TACAR se mantienen estables pero no mejoran.
También en el año 2002, Griffiths et al53 publicaron los resultados de haber tratado a 14 pacientes diagnosticados de ES, con PFR alteradas y cambios en vidrio esmerilado en la TACAR, con 6 pulsos i.v. de metilprednisolona (10 mg/kg) y CF (15 mg/kg). Se observó que esta combinación de fármacos estabilizaba la NI-ES, demostrado con la TACAR, mientras duraba el tratamiento. Los autores recomiendan un tratamiento precoz en los pacientes de riesgo, ya que la DLCO mejoró más en los individuos con enfermedad de duración inferior a los 2 años.
En 2004, Airo et al54 publicaron una serie de 16 pacientes con diagnóstico de NI-ES basado en el LBA y/o TACAR y tratados con pulsos de 750 mg de CF i.v. y 125 mg de prednisolona, cada 3 semanas, durante 6 meses. Tras este período, observaron una discreta mejoría de la CVF y la DLCO, que no alcanzó significación estadística. Los autores realizaron también un metaanálisis en el que se incluyó a 53 pacientes. Basándose en sus resultados y en los procedentes de estudios previos, concluyeron que la CF es útil en la NI-ES y puede mejorar los hallazgos de la TACAR y del LBA, y que su administración i.v. es bien tolerada, con escasos efectos adversos graves.
Kowal-Bielecka et al55 comunicaron en 2005 un análisis retrospectivo de 21 pacientes con patrón en vidrio esmerilado en la TACAR y alteración del LBA o de las PFR, que recibieron CF i.v. durante 6 meses. Observaron que en los pacientes con alveolitis neutrofílica (AN) mejoró la CVF, disminuyó el porcentaje de neutrófilos y eosinófilos en el LBA y se normalizó la PCR. En los pacientes sin AN, la CVF se mantuvo estable, así como el recuento celular en el LBA. Estos datos indican, al igual que en el estudio de Akesson et al49, que la CF es útil, sobre todo, en los pacientes con NI-ES que presentan datos de actividad inflamatoria.
Recientemente se han publicado los resultados del Scleroderma Lung Study, un estudio aleatorizado, multicéntrico, a doble ciego y controlado con placebo en el que se incluyó a 158 pacientes con criterios de alveolitis según el LBA o el TACAR. Los pacientes fueron aleatorizados a recibir CF oral (1-2 mg/kg/día) o placebo durante 1 año y fueron seguidos durante 12 meses más. Un total de 145 pacientes completaron al menos 6 meses de tratamiento. El criterio de valoración principal fue el cambio en la CVF a los 12 meses, y entre los criterios de valoración secundarios se evaluaron la capacidad pulmonar total y la DLCO, el grado de engrosamiento cutáneo y diversos índices de calidad de vida, de actividad y de disnea. Aunque en ambos grupos de tratamiento la CVF empeoró a los 12 meses de seguimiento, el grupo que recibió ciclofosfamida mostró un menor deterioro de la CVF. La CVF en el grupo de ciclofosfamida empeoró una media de 1,0%, frente a un deterioro del 2,6% en el grupo placebo (p < 0,03). Dicha diferencia se mantuvo a los 12 meses de suspender el tratamiento. Por desgracia, el uso de CF no demostró tener ningún efecto en la DLCO, lo que incluso deja abierta la posibilidad teórica de que la CVF se haya deteriorado menos en el grupo de la CF por un efecto no en el parénquima pulmonar, sino en el engrosamiento cutáneo torácico o la función de los músculos implicados en el trabajo respiratorio. La esclerosis cutánea y los índices de disnea, capacidad funcional y calidad de vida, como el SF-36, mejoraron en el grupo de CF, en el que también hubo más efectos secundarios, como la hematuria y la leucocitopenia56.
El trasplante pulmonar es una alternativa en los pacientes con NI-ES evolucionada y sin afección relevante en otros órganos que comprometa la vida. Los resultados del trasplante pulmonar a los 4 años en pacientes con ES son similares a los de los pacientes con FPI, aunque es importante escoger pacientes con NI-ES pero sin otras comorbilidades, como la afección renal, aspiración o afección cutánea muy importante57.
Se han comunicado datos anecdóticos sobre la utilidad del trasplante de células madre en la ES. En algunos casos se ha observado detención e incluso mejoría de la evolución de la enfermedad en sus distintas manifestaciones, incluidas las pulmonares6.
PRONOSTICO
La NI es actualmente la primera causa de morbimortalidad en los pacientes con ES. Un subgrupo de pacientes desarrollará fibrosis pulmonar progresiva y evolucionará hacia una neumopatía restrictiva grave. La media de supervivencia de los pacientes con ES difusa y enfermedad pulmonar es de 78 meses, con una mortalidad a los 8 años del 50%. Se ha comunicado que las razas negra y japonesa, el sexo masculino, la enfermedad inicial y la afección cardíaca se asocian a una neumopatía restrictiva grave6,58.
Los pacientes con NI-ES tienen mejor supervivencia que los pacientes con FPI, incluso tras ajustar por factores de confusión, como la edad de comienzo o la extensión de la enfermedad en la TACAR59. Aunque el patrón histológico más frecuente en la NI-ES es la NINE, la progresión de NINE a NIU podría explicar la mala evolución de algunos pacientes. Por otra parte, Bouros et al30 no pudieron encontrar una mayor supervivencia en los pacientes con ES y NINE respecto a los pacientes con ES y NIU, aunque confirmaron que, en general, la supervivencia a los 5 años era mayor en los pacientes con NI-ES que con FPI.
Diferentes características pueden predecir un pronóstico desfavorable, como un valor basal reducido de la DLCO (< 40% del esperado), un recuento elevado de eosinófilos en el LBA, el deterioro de la capacidad de difusión durante los primeros 3 años de evolución de la ES, la VSG elevada, la hemoglobina baja y la forma difusa de la ES (aunque no la extensión y la gravedad de la afección cutánea). De todos ellos, el más importante es la alteración en la DLCO. La proteinuria podría ser la única variable no pulmonar predictiva de enfermedad pulmonar4,5,58,60.
CONCLUSIONES FINALES
Tras la aparición de los inhibidores de la enzima de conversión de la angiotensina, la afección pulmonar se ha convertido en la primera causa de mortalidad en la ES. Su detección y diagnóstico precoces permiten instaurar el tratamiento antiinflamatorio e inmunosupresor en fases iniciales, cuando los fenómenos inflamatorios activos son susceptibles de modificación por la terapia. Las pruebas de función respiratoria, la difusión de CO, la radiografía y la TACAR de tórax y el análisis del LBA son las técnicas complementarias más útiles en la evaluación de los pacientes. En particular, la TACAR es una técnica no invasiva especialmente valiosa para la detección de la NI-ES, para identificar patrones muy específicos de alveolitis activa y para valorar la respuesta al tratamiento. Se desconoce la terapia óptima de la NI-ES, pero diversos estudios abiertos han indicado que la ciclofosfamida es el inmunosupresor de elección. Los datos extraídos del Scleroderma Lung Study apuntan a una discreta superioridad de la ciclofosfamida oral frente a placebo.