




INTRODUCCION
El síndrome antisintetasa (SA) es un trastorno incluido entre las miopatías inflamatorias idiopáticas que se caracteriza por presentar anticuerpos antisintetasas en el suero. Las diferentes entidades incluidas dentro de las miopatías inflamatorias (dermatomiositis, polimiositis [PM] y miositis por cuerpos de inclusión) son enfermedades autoinmunitarias sistémicas que cursan con debilidad muscular y elevación de enzimas musculares en suero. Las afecciones cutánea, articular y de otros órganos determinan las diferentes presentaciones clínicas de la dermatomiositis o la polimiositis y generalmente no se producen en las miositis por cuerpos de inclusión1. Existen tres grandes rasgos útiles en la clasificación de estas entidades: los hallazgos histopatológicos, la presencia de anticuerpos o la exposición a determinados agentes ambientales2,3, y se ha descrito relación estacional y el desarrollo de enfermedad muscular en pacientes que reciben tratamiento con ciertos fármacos (p. ej., estatinas y fenofibratos), con la aparición de debilidad de la musculatura proximal, elevación de enzimas musculares, anticuerpos antinucleares positivos e incluso verdaderos SA iatrogénicos4,5.
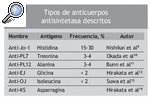
La presencia de los anticuerpos antinucleares es frecuente en las miopatías inflamatorias idiopáticas, y se observa en un 25-35% de los pacientes con PM o dermatomiositis, definiéndose así un síndrome caracterizado por la presencia de los anticuerpos antisintetasa, manos de mecánico, enfermedad pulmonar intersticial, artritis, fiebre y fenómeno de Raynaud, al que se ha llamado SA2. El anticuerpo anti-Jo-1 es el más específico de miositis y el más frecuente en los pacientes con miopatías inflamatorias (20-30%)6 a pesar de que hay evidencia de que la frecuencia de estos anticuerpos es distinta en las diferentes poblaciones etnicogeográficas7. El anticuerpo anti-Jo-1 reacciona con la histidil-ARNt sintetasa, la enzima que cataliza la formación de histidil-ARNt (es decir, la unión covalente del ami noácido histidina a su ARNt afín). Investigar el anti-Jo-1 es fácilmente factible utilizando una inmunoadsorción enzimática (ELISA), inmunoprecipitación de ARN o inmunodifusión de Ouchterlony8. Además, se han descrito anticuerpos contra otras 5 aminoacidil-ARNt sintetasas en pacientes con miopatías inflamatorias idiopáticas (tabla 1).
MANIFESTACIONES CLINICAS
La afección muscular de los pacientes con anticuerpos antisintetasas es similar a la clínica de PM o dermatomiositis sin anticuerpos antisintetasas: inicio subagudo de debilidad muscular proximal, cumpliendo los criterios diagnósticos de Bohan et al15 y respuesta al tratamiento con corticoides e inmunosupresores. Sin embargo, presentan mayor tasa de recurrencia y peor pronóstico16. Hallazgos recientes indican que la patogenia de la miositis en los pacientes con anticuerpos anti-Jo-1 puede ser diferente de la de otras formas de miopatías inflamatorias idiopáticas, al encontrar mayor fragmentación del tejido conectivo perimisial sin mostrar marcada pérdida de densidad capilar, presente por ejemplo en la dermatomiositis17.
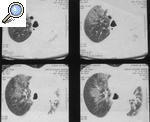
El hallazgo clínico más relevante del SA es la afección pulmonar intersticial y su potencial impacto en el pronóstico de la enfermedad. Puede presentarse de forma fulminante como disnea aguda18, como neumonía intersticial no específica (NINE)19 o como bronquiolitis obliterante con neumonía organizada (BONO)20 (fig. 1). Es poco frecuente que la afección pulmonar preceda a la miositis, pero en algunos pacientes la miopatía puede ser subclínica o no ocurrir nunca y el SA puede manifestarse únicamente por la enfermedad pulmonar intersti-cial21-23. Por ello, la determinación de los anticuerpos antisintetasas es de utilidad en el abordaje diagnóstico de las neumopatías intersticiales idiopáticas, lo que permite mejorar el pronóstico de estas enfermedades24,25.
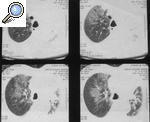
Figura 1
La afección articular del SA se caracteriza por ser poliarticular y simétrica, e incluso la forma inicial de la enfermedad. Es no erosiva, pero puede producir subluxaciones y asociarse a calcinosis periarticular26,27.
Las denominadas "manos de mecánico", caracterizadas por hiperqueratosis con fisuras en los bordes de los dedos, no son específicas de los pacientes con antisintetasas y se las ha visto asociadas a los anticuerpos anti-PM-Scl y anti-Mi-228,29.
También es más frecuente en los pacientes con SA, fenómeno de Raynaud y fiebre durante la fase activa de la enfermedad8 (tablas 2 y 3).


Se ha sugerido oscilaciones en el título de anticuerpos anti-Jo-1 con la actividad de la enfermedad, pero no se ha demostrado la utilidad clínica de monitorizar los títulos, pues el anticuerpo persiste a pesar de la remisión de la clínica31. La determinación de los anticuerpos anti-Jo-1 permite la confirmación diagnóstica (por su alta especificidad) y su capacidad de predecir manifestaciones clínicas (miositis o afección pulmonar) y pronósticas, aunque en algunos casos hay pacientes que presentan similitudes clínicas y anticuerpos anti-PM-Scl o anti-U1RNP32,33.
PRONOSTICO
Algunas características demográficas, clínicas y serológicas de las miopatías inflamatorias idiopáticas se han asociado a peor pronóstico: la edad avanzada al diagnóstico, la presencia de miositis grave, el retraso en el inicio del tratamiento, la afección cardíaca, pulmonar o gastrointestinal y la presencia de neoplasia, la miositis por cuerpos de inclusión o la asociación de los anticuerpos antisintetasas2. El tratamiento con glucocorticoides puede ser eficaz en la afección muscular, pero en la enfermedad pulmonar no es suficiente y puede progresar a pesar del tratamiento inmunosupresor34. En diversos trabajos, se ha descrito que la asociación del SA con el anticuerpo anti-Ro puede predisponer al desarrollo de una forma más grave de enfermedad pulmonar intersticial35. La afección muscular tiene peor pronóstico en los pacientes con PM y anticuerpos antisintetasa que en los pacientes sin estos anticuerpos, y la afección pulmonar de este síndrome tiene mejor pronóstico que la fibrosis pulmonar idiopática36,37. Los agentes inmunosupresores deben ser utilizados en el tratamiento del SA fundamentalmente cuando hay enfermedad pulmonar intersticial, en la afección articular resistente o en la recidiva de la enfermedad al disminuir la dosis esteroidea34,38. Spath et al34 demostraron en 2004 que la remisión con tratamiento esteroideo se logra únicamente en el 25-68% de los pacientes y las recaídas después de la remisión completa varían en un 6-43%, y son más frecuentes las recidivas tardías después de la remisión en el SA que en otro tipo de miopatías34.
TRATAMIENTO
El tratamiento del SA depende fundamentalmente de la afección pulmonar intersticial, requiriendo altas dosis de glucocorticoides intravenosos u orales e inmunosupresores39-41. El tratamiento fisioterápico o rehabilitador es importante para mantener y mejorar la función muscular y articular41. Es necesario además un tratamiento multidisciplinario en el que colaboren reumatólogos, neumólogos, dermatólogos, psicólogos y rehabilitadores.
El agente inmunosupresor más frecuentemente utilizado en la afección pulmonar del SA es la ciclofosfamida, utilizada sobre todo en forma de pulsos intravenosos42, con mejoría demostrable a los 6 meses de tratamiento en las pruebas funcionales respiratorias y en los hallazgos radiológicos (TACAR)43. En otras ocasiones se ha utilizado de forma oral o asociado a otros agentes como azatioprina o ciclosporina A44-46. El tacrolimus también se ha utilizado en la afección pulmonar intersticial asociada al SA47.
Aunque los glucocorticoides son el tratamiento inicial para tratar la miositis, ocasionalmente es necesario el uso de metotrexato en dosis de 7,5-20 mg semanales vía oral o parenteral, para control de la actividad muscular o articular, solo o asociado a otros agentes como la ciclosporina A48. Las inmunoglobulinas intravenosas se han utilizado en casos de PM y dermatomiositis resistentes a tratamientos habituales a dosis de 1 mg/kg/día durante 2 días por mes, durante 4-6 meses49,50 y en otras ocasiones asociadas a ciclosporina A51,52. Azatioprina53 o micofenolato mofetilo54 son otros de los agentes utilizados para tratamiento de la afección muscular o en la enfermedad intersticial pulmonar del SA.
La leflunomida también se ha utilizado como tratamiento del SA en casos de afección articular resistentes a otros tratamientos habituales55.
Las terapias biológicas utilizadas en varias enfermedades autoinmunitarias resistentes a los tratamientos habituales (artritis reumatoide, lupus eritematoso sistémico, síndrome de Sjögren primario, vasculitis, SA y sarcoidosis, entre otras) se han usado en miopatías inflamatorias y en el SA con afección muscular resistente, con buena tolerancia y eficacia demostrada a corto plazo: así, la neutralización del factor de necrosis tumoral alfa puede ser una opción terapéu-tica, pues se ha demostrado que esta citocina está implicada en la patogenia de la enfermedad. Aún son pocos los casos tratados, pero se ha demostrado eficacia con infliximab56, etanercept57 y adalimumab58. El anticuerpo monoclonal anti-CD20 rituximab, en dosis de 1 g los días 0 y 14, ha sido utilizado en miositis resistentes a tratamientos convencionales59-61.
Otros tratamientos utilizados han sido el trasplan-te de células autólogas, utilizado en la afección intersticial pulmonar rápidamente progresiva en combinación con altas dosis de ciclofosfamida y corticoides62 y en modelos animales murinos, la inhibición de la fractalcina (citocina expresada en células mononucleares y endoteliales) ha conse-guido mejoría de los parámetros de afección muscular63.
CONCLUSIONES
El interés del SA radica en la asociación con un cuadro de afección pulmonar intersticial difusa en el contexto de las miopatías inflamatorias idiopáticas. Por tanto, en el abordaje diagnóstico de estas enfermedades y de las neumopatías intersticiales idiopáticas es necesario sistematizar la determinación de los anticuerpos antisintetasas para establecer un diagnóstico y un tratamiento adecuados y precoces que permitan mejorar el pronóstico de estas entidades