


Muchas veces el impedimento estérico es suficiente para explicar los diferentes rendimientos de los isómeros obtenidos en las reacciones de sustitución electrofílica aromática. Sin embargo, esto es insuficiente cuando las diferencias en los rendimientos de reacción son muy marcadas. Lo último se debe, por lo general, a que intervienen otros factores, aparte del estérico, los cuales son de carácter eléctrico, por ejemplo, rechazo por parte de cargas eléctricas del mismo signo entre los reactivos. Por lo anterior, un factor muy importante a tomar en cuenta es el impedimento eléctrico, un concepto teórico nuevo. Se ha tomado como ejemplo de lo anterior la nitración de la 2-aminopiridina y se da una explicación teórica amplia, tanto de la regioquímica observada como de los rendimientos de reacción de los isómeros nitrados obtenidos, tomando en cuenta también otros factores como los momentos dipolo.
Además, se discute el tipo de transposición que se lleva a cabo, en medio ácido, en la 2-nitraminopiridina. Nuestra discusión teórica concuerda con los resultados experimentales de trans-nitración que han sido descritos y se confirmó al contrastarla con los resultados obtenidos en experimentos de termólisis y de fotólisis de la 2-nitraminopiridina.
The different isomer yields observed in many aromatic electrophilic substitution reactions can be explained by steric hindrance. However, this is not the case when there are drastic differences in the reaction yields of the isomeric products obtained. This is generally due to the presence of other factors, for instance, electric rejection between two positive charges in the reaction stage. Thus, a very important point to bear in mind is electric hindrance, a new theoretical concept. We have taken as an example 2-aminopyridine nitration. We provide an extended theory on this subject, which is in accordance with the observed regiochemistry and with the reaction yields of the isomeric products obtained. Dipole moments were also taken into account.
We discuss too the 2-nitraminopyridine rearrangement in acidic medium. The theoretical discussion is also in agreement with reported trans-nitration experimental results. Our proposals were also contrasted with the findings from thermolysis and photolysis carried out with 2- nitraminopyridine.
Impedimento eléctrico (Electric Hindrance) es un concepto nuevo en Química Orgánica, propuesto por nosotros, y reconocido por la American Chemical Society1. En la presente comunicación se utiliza este concepto teórico para explicar la diferencia notable existente en los porcentajes de los isómeros obtenidos en la nitración de la 2-aminopiridina. Este concepto teórico se ha empleado en unión de otros conceptos y conocimientos teóricos de uso corriente en Química Orgánica, teniendo en cuenta los datos experimentales de los diferentes rendimientos de reacción, con el fin de establecer una relación causal.
En este estudio se realizó un análisis razonado de los diferentes efectos eléctricos y electrónicos presentes tanto en el sustrato como en los intermediarios de reacción involucrados en la nitración de la 2-aminopiridina y en la transformación de la 2-nitraminopiridina (2-piridilnitramina) en compuestos nitrados directamente en el anillo.
Por otra parte, nuestra propuesta teórica respecto al tipo de la transposición que se lleva a cabo en la 2-nitraminopiridina, coincide con las conclusiones de otros autores, basadas en los datos obtenidos en experimentos de trans-nitración, como se verá en la Parte Teórica.
Parte teóricaSe han efectuado varias revisiones de la química de las aminopiridinas2-6. Una reacción interesante es la nitración de la 2-aminopiridina debido a los contrastes que presenta su regioquímica.
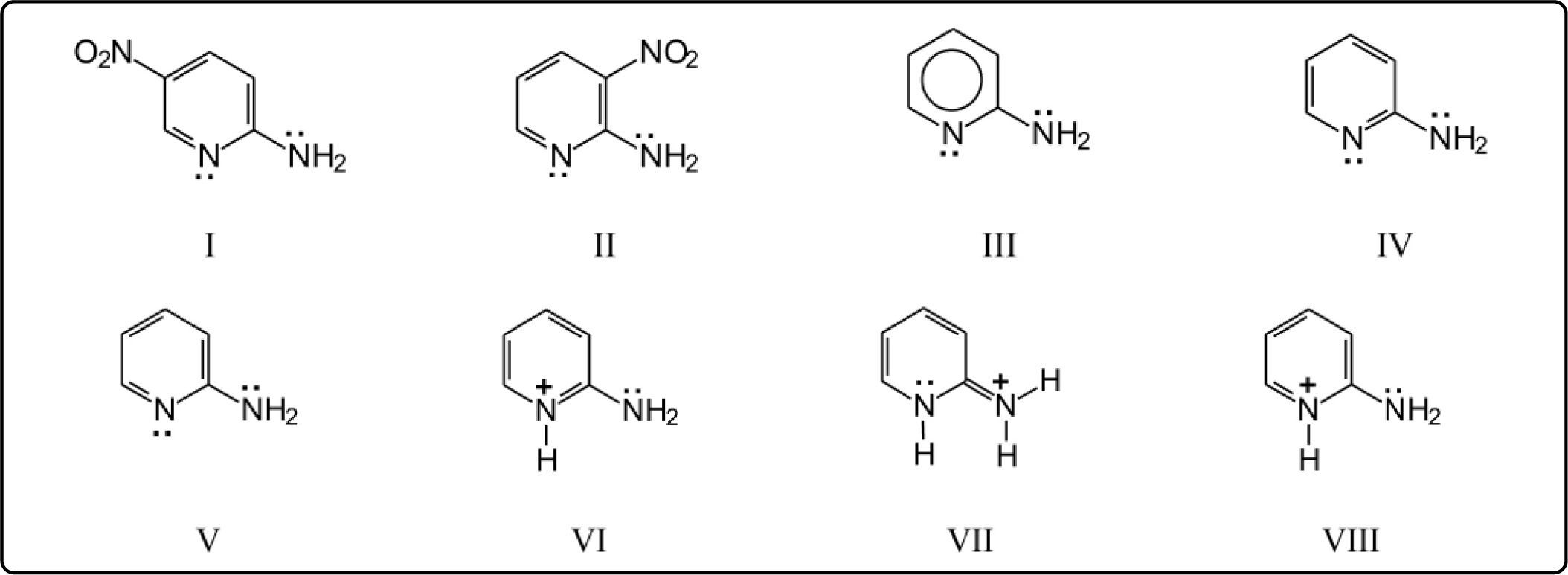
Tchitchibabin7,8 nitró la 2-aminopiridina, obteniendo 2-amino-5-nitropiridina como producto principal, I, y en cantidad mucho menor, 2-amino-3-nitropiridina, II. Dado que los trabajos de Tchitchibabin fueron publicados en ruso, y los resúmenes en inglés y en francés son cortos, investigadores posteriores, ingleses9 y americanos10, basados en los experimentos originales, publicaron métodos detallados de la nitración de la 2-aminopiridina.
La 2-amino-3-nitropiridina, II, es arrastrable por vapor de agua. Esto ha sido explicado posteriormente11, debido a la formación de puentes de hidrógeno intramoleculares entre los grupos amino y nitro en posición orto. Este quelato impide la formación de puentes de hidrógeno intermoleculares, caso que ocurre en la 2-amino-5-nitropiridina, evitando su volatilidad. La relación en peso entre los isómeros obtenidos es de 9 a 1. También se ha descrito la separación de las dos aminonitropiridinas por sublimación11,12, además de las separaciones originales por cristalización y por arrastre con vapor de agua.
Debido a que no se ha dado una explicación teórica de la preponderancia del isómero 5-nitro y, sobre todo, a la marcada desproporción de los rendimientos, consideramos de interés proponer una teoría que explique la regioquímica tan especial de esta reacción.
Obviamente, la formulación bianular (anillo atómico y anillo electrónico) de la 2-aminopiridina, III, no es favorable, ya que da la idea de una reactividad igual en todas las posiciones sustituibles. De hecho, hay heteronomía, no isonomía. De las 2 estructuras canónicas, IV y V, la IV no permite explicar las reacciones de sustitución en el anillo, pues al protonarse el nitrógeno endocíclico, debido al medio ácido, queda un agrupamiento imonio-enamina, VI, que atraería el par electrónico del NH2, VII, perdiéndose la contribución electrónica a orto y para, lo cual no es así, ya que la reactividad de la 2-aminopiridina7 es mucho mayor que la de la piridina. Por lo anterior, las estructuras favorecidas son la V y la VIII.
En las estructuras anteriores, no se ha tenido en cuenta la protonación del nitrógeno exocíclico, no porque no ocurra, sino porque los resultados experimentales indican que el grupo amino facilita enormemente esta nitración y, por tanto, que la formación de la sal de amonio es reversible.
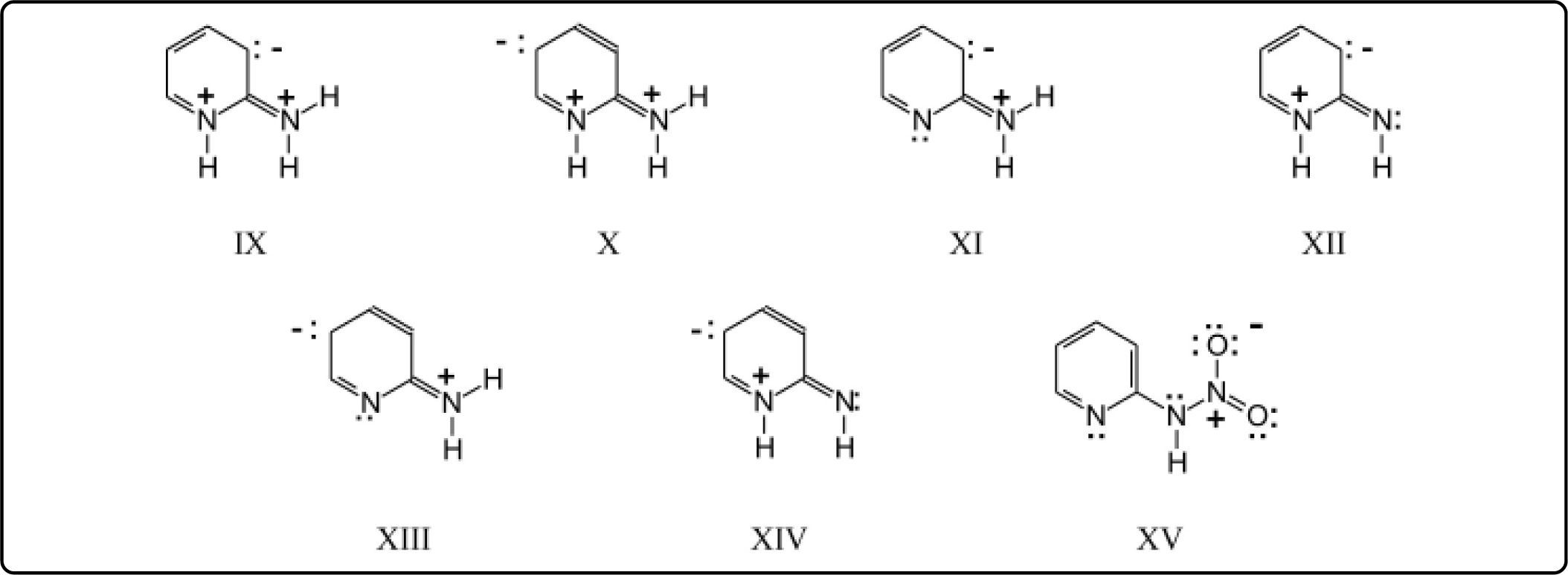
Consideremos ahora las estructuras resonantes de la fórmula VIII: en la estructura IX, el grupo amino ha formado un dipolo 1,3, mientras que en la X se tiene una estructura semiquinoide y el dipolo formado por el grupo amino es 1,5. Queda, además, en ambas estructuras IX y X, otro dipolo 1,3 entre los nitrógenos con carga positiva. Esta cercanía de cargas positivas puede eliminarse por desprotonación. Se pueden tener los siguientes casos: XI-XIV. En las estructuras XII y XIV, con grupos imino exocíclicos, se tienen dipolos 1,3, lo cual haría muy similar la reactividad de estos 2 intermediarios. En cambio, los isómeros XI y XIII, con dipolos 1,3 y 1,5, respectivamente, tendrían reactividades muy diferentes. Estas estructuras derivan directamente de la 2-aminopiridina sin protonar. Es decir, nos indican que la reactividad de este compuesto está regida por el tipo de dipolo que el grupo amino forma.
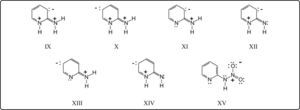
El ión nitronio, al acercarse al carbanión de la estructura XI, encuentra cierto impedimento estérico, pero sobre todo, repulsión eléctrica por la carga positiva del nitrógeno. De manera que para formar el isómero nitrado en 3 se debe sobrepasar este impedimento eléctrico. En el caso del carbanión de la estructura XIII, se puede ver que el ión nitronio no encuentra repulsión eléctrica y, por lo mismo, el compuesto nitrado en la posición 5 es el altamente favorecido.
Por otra parte, si consideramos las atracciones eléctricas existentes en los intermediarios XI y XIII, se ve que el momento dipolo es menor en XI que en XIII. XI es el intermediario de menor energía, más cercano a la estructura basal, neutra. En cambio, XIII es el intermediario de más alta energía, más inestable y, por lo mismo, más reactivo. Otra consideración que puede hacerse es que el carbanión en 5 (XIII), más alejado de la carga positiva, está más aislado (carbanión desnudo), más disponible que el carbanión en 3 (XI). Estas últimas consideraciones están de acuerdo con los resultados experimentales13,14 que indican que la proporción de 5-nitro-2-aminopiridina es mayor al aumentar la temperatura de reacción, es decir, al favorecer la formación del intermediario de mayor energía, XIII.
Tchitchibabin13,14 también dio a conocer que cuando se agrega ácido nítrico a una solución al 10% de 2-aminopiridina en H2SO4 concentrado, fuertemente enfriada, se obtiene la 2-nitraminopiridina, XV. Es un compuesto anfótero, soluble en ácidos minerales y en soluciones alcalinas débiles. Las soluciones alcalinas concentradas precipitan las sales alcalinas de la nitramina (nitronatos). La solución de carbonato sódico disuelve la 2-nitraminopiridina, con desprendimiento de anhídrido carbónico.
La explicación teórica de la formación de la 2-nitraminopiridina es que la temperatura baja, menor de 40° C, no favorece el aporte electrónico del nitrógeno exocíclico al anillo y el disturbio de la aromaticidad. Por lo anterior, el ión nitronio se fija en el par electrónico libre del nitrógeno exocíclico, formándose por desprotonación la 2-nitraminopiridina. En este caso el producto resultante es el cinético, y no el termodinámico, como cuando la temperatura es mayor de 40° C.
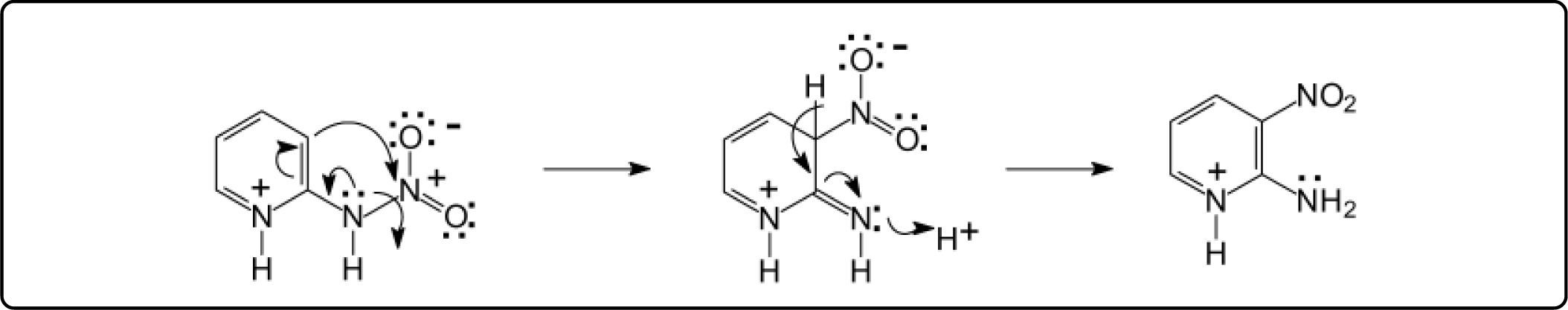
La 2-nitramino-piridina, disuelta en H2SO4 y calentada a 50° C o más, se transforma en los productos de nitración anular, los isómeros 2-amino-3nitro- y 2-amino-5-nitropiridina2. Se ha pensado en una posible migración 1,3 del grupo nitro6, para formar la 2-amino-3-nitropiridina. Sin embargo, esto no es posible debido a razones teóricas y porque contradice resultados experimentales. Si así fuera, se requiere la entrada al anillo del par electrónico del amígeno del grupo nitramino, como se indica en el Esquema 1.

Es problemático que este par de electrones pueda alterar la aromaticidad, teniendo en cuenta que el nitrógeno está unido a un grupo electrocaptor, el nitro. Además, la migración 1,3 del grupo nitro implica un mecanismo cíclico concertado de 4 centros, el cual es poco probable, ya que son favorecidos los de 5 y 6 centros.
Por otra parte, de ocurrir una migración 1,3 del grupo nitro, el compuesto resultante, la 2-amino-3-nitropiridina, sería no sólo el producto principal, sino el único obtenido, ya que no se ve motivo para la migración del grupo nitro de la posición 3 a la 5, no contigua. Sin embargo, los resultados experimentales son los mismos que cuando se hace la nitración directa de la 2-amino-piridina, siendo el isómero 2-amino-5-nitro el altamente favorecido.
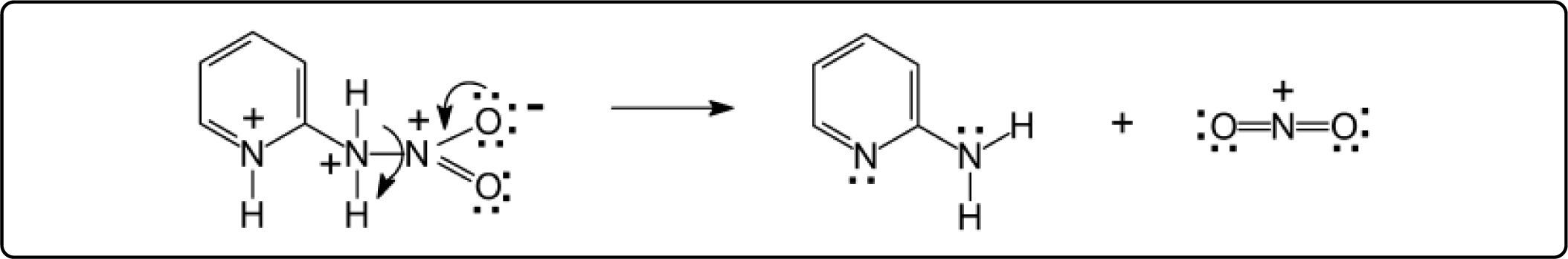
Una alternativa mucho más viable que el mecanismo anterior, es la protonación del nitrógeno exocíclico debido al medio fuertemente ácido (H2SO4) y a la temperatura más alta. El intermediario resultante, es de alta energía debido a la existencia de 2 cargas eléctricas positivas contiguas. Esta repulsión origina la separación del grupo NO2, en forma de ión nitronio, y la regeneración de la 2-aminopiridina, como se indica en el Esquema 2.

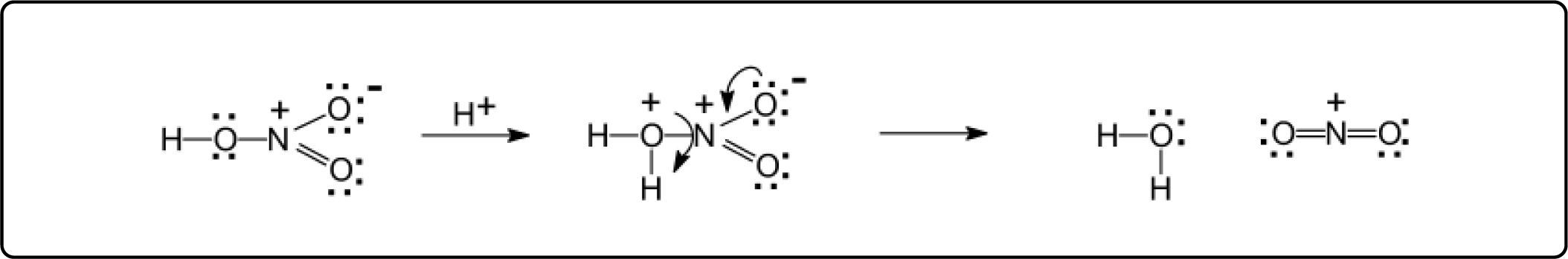
De esta manera, se tiene la misma reactividad que cuando se lleva a cabo la nitración directa y a temperatura mayor a 40° C, obteniendo los mismos rendimientos de los 2 isómeros en ambos casos. La disociación propuesta es similar a la reacción de formación del ión nitronio a partir de ácido nítrico, Esquema 3.

Nuestras deducciones teóricas respecto a la naturaleza y al curso de esta transposición se confirman al contrastarlas con los siguientes resultados experimentales:
Experimentos de trans-nitración (nitración cruzada, crossover experiments)15,16, con acetanilida como compuesto adicional, están de acuerdo con un mecanismo de transposición intermolecular. Esto despeja las dudas respecto al tipo de transposición, provenientes de intentos de intercambio de 15N empleando Na15NO317. Dado que el nitrato no es la especie reactiva, la dificultad de formar un ión 15NO2+, en las condiciones de transposición, introduce un factor extraño a la transposición en sí.
Por otra parte, se han efectuado experimentos en diferentes concentraciones de ácido sulfúrico. Los resultados indican que los rendimientos de reacción decrecen al aumentar la dilución de la solución18. Esto está de acuerdo con el primer paso de nuestro mecanismo: un medio fuertemente ácido favorece la protonación del amígeno de la nitramina, generando un intermediario de alta energía (2 cargas positivas en átomos contiguos), lo cual induce la separación de un ión nitronio (disociación por acidólisis).
La transposición de la 2-nitraminopiridina también se ha llevado a cabo por termólisis y por fotólisis. Dado que estos métodos no involucran iones sino radicales libres, la información relacionada con estos experimentos es indirecta pero interesante. La transposición por termólisis19, llevada a cabo en clorobenceno a 132°C, sin catálisis ácida, genera los mismos regioisómeros, pero los rendimientos de reacción están invertidos. El producto mayoritario es ahora la 3-nitro-2-aminopiridina (40%) y el minoritario la 5-nitro-2-aminopiridina (26%). La transposición puede llevarse a cabo en soluciones de anisol o de m-xileno, sin nitración cruzada. Por lo tanto, la migración del grupo nitro es intramolecular, en contraste con el mecanismo intermolecular que ocurre en medio ácido que hemos propuesto y confirmado por los experimentos de trans-nitración.
En el experimento por fotólisis20, la irradiación de la 2-nitraminopiridina con lámpara de mercurio origina 2-amino-3-nitro- y 2-amino-5-nitro-piridinas. La proporción es de 6.26 : 1, al contrario de los resultados obtenidos en la transposición catalizada por ácido. El compuesto ahora mayoritario, la 2-amino-3-nitropiridina, es precursor de la 2,3-diaminopiridina, útil para condensaciones orto conducentes a la formación de poliazaheterociclos fusionados. Sin embargo, el rendimiento total de ambos productos es de solamente 12%, lo cual descarta esta ruta como método preparativo. Es de hacer notar que en esta transposición vía radicales libres, el regioisómero favorecido es el resultante de una migración vicinal, muy probablemente mediante un mecanismo de reacción intramolecular, a diferencia de la transposición en medio ácido, en la cual el isómero mayoritario tiene el grupo nitro en la posición más alejada al grupo nitramino original.
En la transposición de la 2-nitraminopiridina hay similitud con la transposición de cloraminas y nitrosaminas aromáticas21, en que las tres proceden vía un mecanismo intermolecular. Sin embargo, hay notorias diferencias: con las cloraminas y con las nitrosaminas se utiliza exclusivamente HCl; con las cloraminas la especie reactiva es neutra (Cl2), a diferencia del ión nitronio en el caso de la 2-nitraminopiridina. Con las nitrosaminas el primer paso de reacción es la formación de una amina secundaria y cloruro de nitrosilo, en vez de un ión nitrosonio formal.
ConclusionesSe proporcionó una teoría que explique las experiencias factuales, es decir, la regioquímica y los rendimientos de reacción observados en la nitración de la 2-aminopiridina. Nuestro estudio incluye dilucidar y discutir el mecanismo de la transposición en medio ácido de la 2-nitraminopiridina en compuestos nitrados en el núcleo piridínico. El interés e importancia radican en encontrar el fundamento de las reacciones químicas estudiadas (frontera epistemológica). Esta interpretación de resultados tiene importancia tanto en Educación Química como en Química Teórica.
Los puntos sobresalientes de este estudio son:
- •
El impedimento estérico no es el factor determinante de la regioquímica en la nitración de la 2-aminopiridina.
- •
El impedimento eléctrico, debido al rechazo entre cargas positivas, es el factor que rige la regioquímica en la reacción anterior.
- •
La 2-nitraminopiridina es el producto cinético de reacción al nitrar la 2-aminopiridina.
- •
Los isómeros resultantes de la nitración anular son los productos termodinámicos de reacción.
- •
La isomerización de la 2-nitraminopiridina a los productos de nitración anular no ocurre mediante un mecanismo cíclico concertado de 4 miembros, como tentativamente se ha propuesto.
- •
La isomerización anterior ocurre mediante la secuencia: protonación-disociación-nitración anular.