


El benzo[a]pireno (b[a]p), es un hidrocarburo aromático policíclico (PAH) producto de combustiones incompletas de materia orgánica. Es considerado como carcinógeno debido a que los metabolitos derivados de su biotransformación, como el benzo [a] pireno diol epóxido (b[a]pDE), tienen propiedades mutagénicas y carcinogénicas. El b[a]p es metabolizado por las enzimas citocromos P450 1A1 (CYP1A1) y el citocromo P450 1B1 (CYP1B1) cuya expresión es inducida por la vía de señalización del receptor de hidrocarburos de arilo (AhR). Algunos estudios sugieren que el AhR es capaz de modular otros procesos celulares, además de la desintoxicación de xenobióticos como el desarrollo, diferenciación, proliferación, respuesta inmune, promoción del cáncer y apoptosis. Esto mediante la modulación de vías de señalización mediadas por proteínas cinasas como la del receptor del factor de crecimiento epidermal (EGFR, por sus siglas en inglés). Sin embargo, no es clara la función del AhR en estos procesos ni cómo el AhR es capaz de interactuar con otras vías de señalización. Las alteraciones celulares inducidas por b[a]p son complejas y pueden estar mediadas por más de una vía de señalización y la activación de múltiples genes, por lo que es esencial saber qué vías están involucradas en el metabolismo de los PAH para una mejor comprensión de las bases moleculares de enfermedades como el cáncer.
Benzo[a]pyrene (b[a]p), is a polycyclic aromatic hydrocarbon (PAH) product of incomplete combustion of organic matter. B[a]p is considered as a carcinogen due to the mutagenic and carcinogenic properties of its biotransformation derivatives, such as benzo[a]pyrene diol epoxide (b[a]pDE). B[a]p is metabolized by cytochrome P450 enzymes 1A1 (CYP1A1) and 1B1 (CYP1B1), whose expression is regulated by the aryl hydrocarbon receptor (AhR) signaling pathway. Studies suggest that AhR is involved in the regulation of other cellular processes, in addition to detoxification of xenobiotics, such as development, differentiation, proliferation, immune response, cancer development, and apoptosis, by modulating signaling pathways mediated by protein kinases such as the epidermal growth factor (EGFR). However it is not clear the role of AhR in these processes nor the mechanisms of AhR interaction with other signaling pathways. Cellular changes induced by b[a]p are complex and may be mediated by more than one signaling pathway and by the activation of multiple genes, so it is essential to know which pathways are involved in the metabolism of PAHs for a better understanding of the molecular basis of diseases such as cancer.
Los hidrocarburos aromáticos policíclicos (PAH, por sus siglas en inglés) son compuestos químicos conformados por dos o más anillos aromáticos fusionados. Son producto de combustiones incompletas de combustibles fósiles, madera, carbón y tabaco, principalmente. Son componentes importantes del material particulado de 2.5 micras y 10 micras de tamaño (PM 2.5 y PM 10, respectivamente) los cuales se han relacionado con alteraciones respiratorias. El benzo[a]pireno (b[a]p) es un PAH considerado como carcinógeno y está relacionado con la etiología de cáncer de pulmón1. Este compuesto es metabolizado mediante la vía de señalización del receptor de hidrocarburos de arilo (AhR, por sus siglas en inglés). Los efectos carcinogénicos del b[a]p pueden ser explicados, en términos generales, por dos mecanismos: la vía genómica y la vía no genómica. La primera consiste en la inducción de genes que codifican enzimas que participan en la bioactivación del b[a]p. Por ejemplo, en presencia de b[a]p la enzima citocromo P450 1A1 (CYP1A1) produce dioles epóxidos del b[a]p que son capaces de reaccionar con el ADN produciendo lesiones premutagénicas. Este mecanismo explica las observaciones de Vogelstein y cols. en donde células tumorales de pulmón presentan diez veces más mutaciones que otros tipos de tumores2. El segundo mecanismo tiene que ver con la participación del AhR en la estimulación, promoción y progresión de tumores, mediante la alteración de las vías de señalización relacionadas con la proliferación celular y del sistema inmune. Es probable que estos efectos estén mediados por la intercomunicación de la vía genómica y la no genómica. Sin embargo, el papel del AhR en esta intercomunicación no es muy conocido. No obstante, existen trabajos que podrían ayudar a comprender su papel en la producción de tumores, destacando aquellos donde se sugiere la interacción de la vía de señalización del AhR con la vía de receptores tipo ErbB, como el receptor del factor de crecimiento epidermal (EGFR). Por lo que el presente trabajo se enfoca en revisar los trabajos que muestran evidencias de la intercomunicación entre estas dos vías de señalización inducidas bajo el estímulo de los PAH.
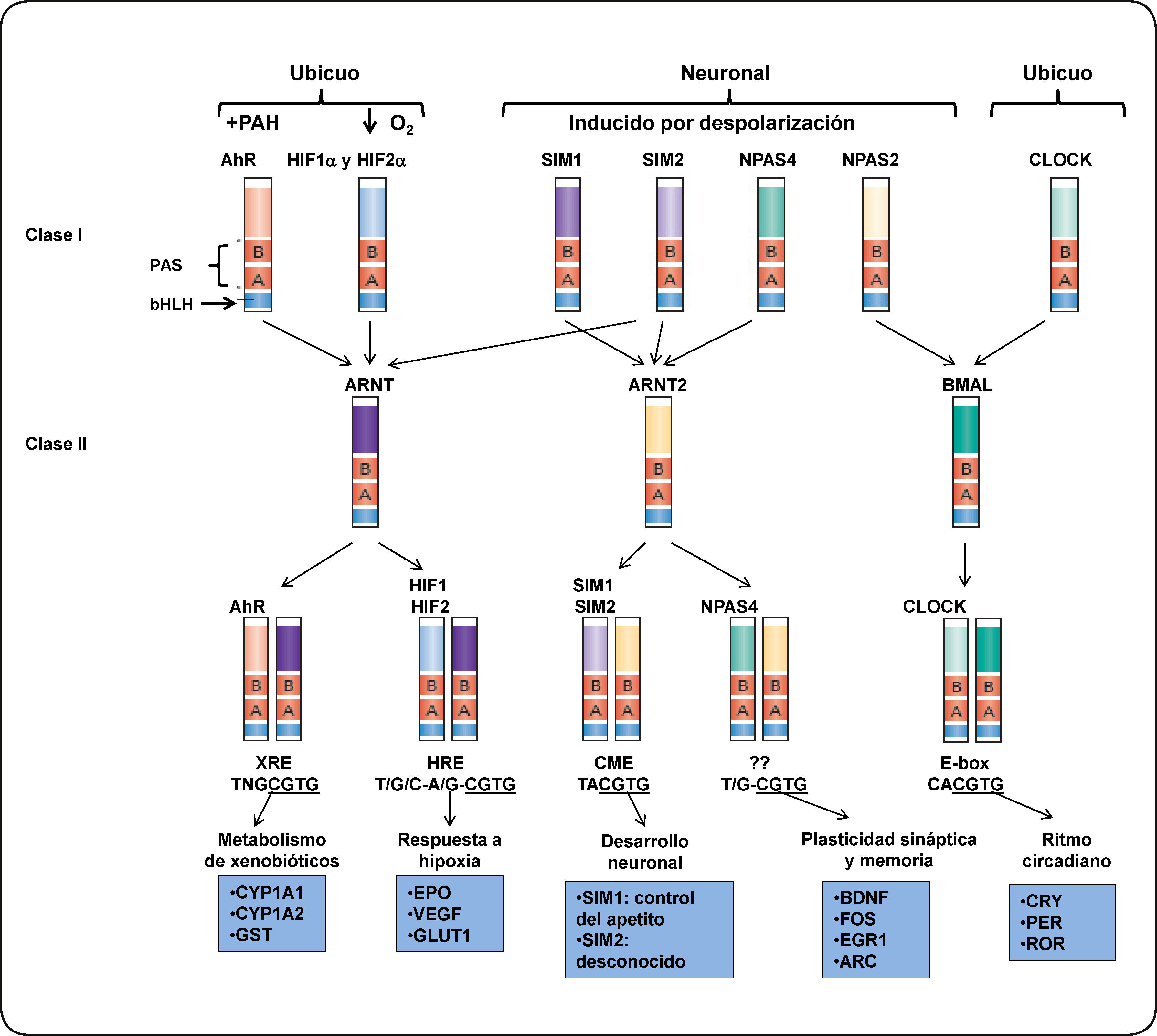
EL RECEPTOR DE HIDROCARBUROS DE ARILO (AHR)El receptor de hidrocarburos de arilos (AhR) es un factor de transcripción citoplasmático perteneciente a la familia de los factores de transcripción con dominio bHLH-PAS (basic helix loop helix-PER ARNT-SIM).3,4 Esta familia de proteínas desempeña un papel importante en diversos procesos celulares como desarrollo, adaptación a hipoxia, control del ciclo circadiano y metabolismo de xenobióticos5. Estas proteínas se dividen en dos clases: Clase I (α) y clase II (β). La expresión de las proteínas de la clase I, generalmente está restringida a un tejido o es regulada por una señal y forman heterodímeros sólo con las proteínas de la clase II, mientras que la clase II, es ubicua o su expresión no está sometida a regulación y puede formar homodímeros con proteínas de su misma clase.6 Por ejemplo, la expresión del AhR o el factor inducible por hipoxia alfa (HIF-α), en humano, es inducible. Estas proteínas dimerizan con el translocador nuclear del receptor de arilos (ARNT, por sus siglas en inglés), el cual, por lo general es ubicuo o su expresión no está sometida a regulación (Figura 1).
. La expresión de los factores de la clase I posiblemente está restringida a un tejido (como la proteína neuronal con dominio PAS (NPAS)) o se active en respuesta de un estímulo, como el AhR. Los heterodímeros se unen a secuencias específicas en el ADN y regulan la expresión de genes blanco. De este modo, son capaces de controlar diversos procesos celulares en el desarrollo, homeostasis celular o en procesos celulares en respuesta a un estrés fisiológico. Los factores de transcripción pertenecientes a la clase II son capaces de formar dímeros con más de un factor de la clase I aunque algunas combinaciones pueden ser limitadas in vivo. Por ejemplo, la expresión del factor ARNT2 está restringida a neuronas y es funcional sólo cuando dimeriza con el factor SIM1. Otro ejemplo es el AhR, este factor unido a un ligando puede dimerizar con ARNT o ARNT2 in vitro. Sin embargo, sólo los dímeros AhR/ARNT pueden activar la transcripción de sus genes blanco32. Elementos que se muestran en la figura: ARC, proteína asociada al citoesqueleto regulada por actividad; BDNF, factor neurotrófico derivado del cerebro; CME, elemento central de la línea media; CLOCK (por sus siglas en inglés), Circadian Locomotor Output Cycles Kaput; CRY, criptocromos; CYP1A1, citocromo P4501A1; EGR1, proteína de respuesta de crecimiento temprano 1; EPO, eritropoyetina; GLUT 1, transportador de glucosa 1; GST, glutatión S-transferasa; HRE, elemento de respuesta a hipoxia; PER, proteína homóloga del ciclo circadiano; ROR, receptor huérfano relacionado al ácido retinoico; SIM proteína single-minded; VEGF, factor de crecimiento endotelio vascular; XRE, elemento de respuesta a xenobióticos. Modificado de Bersten et al.33.")
Estructura y función de las proteínas de la clase I y II de la familia bHLH-PAS. Se muestran ejemplos de los factores de transcripción heterodiméricos bHLH-PAS. Los dímeros son formados por la interacción entre los factores de la clase I y los factores de la clase II (como lo indican las flechas). La expresión de los factores de la clase I posiblemente está restringida a un tejido (como la proteína neuronal con dominio PAS (NPAS)) o se active en respuesta de un estímulo, como el AhR. Los heterodímeros se unen a secuencias específicas en el ADN y regulan la expresión de genes blanco. De este modo, son capaces de controlar diversos procesos celulares en el desarrollo, homeostasis celular o en procesos celulares en respuesta a un estrés fisiológico. Los factores de transcripción pertenecientes a la clase II son capaces de formar dímeros con más de un factor de la clase I aunque algunas combinaciones pueden ser limitadas in vivo. Por ejemplo, la expresión del factor ARNT2 está restringida a neuronas y es funcional sólo cuando dimeriza con el factor SIM1. Otro ejemplo es el AhR, este factor unido a un ligando puede dimerizar con ARNT o ARNT2 in vitro. Sin embargo, sólo los dímeros AhR/ARNT pueden activar la transcripción de sus genes blanco32. Elementos que se muestran en la figura: ARC, proteína asociada al citoesqueleto regulada por actividad; BDNF, factor neurotrófico derivado del cerebro; CME, elemento central de la línea media; CLOCK (por sus siglas en inglés), Circadian Locomotor Output Cycles Kaput; CRY, criptocromos; CYP1A1, citocromo P4501A1; EGR1, proteína de respuesta de crecimiento temprano 1; EPO, eritropoyetina; GLUT 1, transportador de glucosa 1; GST, glutatión S-transferasa; HRE, elemento de respuesta a hipoxia; PER, proteína homóloga del ciclo circadiano; ROR, receptor huérfano relacionado al ácido retinoico; SIM proteína single-minded; VEGF, factor de crecimiento endotelio vascular; XRE, elemento de respuesta a xenobióticos. Modificado de Bersten et al.33.
El AhR fue uno de los primeros factores de transcripción perteneciente a la familia bHLH en ser descubierto y estudiado. Su cADN fue clonado por primera vez por Ema et al.7 en 1992 y por Burbach et al.8 en el mismo año. El gen fue caracterizado por Schmidt et al.9 y desde entonces se han clonado homólogos del AhR de diferentes especies; por ejemplo, en humano (Homo sapiens)10, ratón (Mus musculus)7,8,11,12, rata (Rattus norvergicus; Sprague-Dawley)13, Hamster (Mesocrisetus auratus)14, delfín (Lagenorhynchus auratus)15 y foca (phoca sibirica)16. También han sido clonadas secuencias de vertebrados no mamíferos como aves17,18, anfibios18, peces cartilaginosos19,20, lampreas19 e invertebrados como bivalvos (Mya arenaria)21, nematodos (Caenorhabditis elegans)19 y la mosca de la fruta (Drosophila melanogaster)22. El gen que codifica para el AhR en humano se localiza en el brazo corto del cromosoma 7 en la posición 1523 y se expresa principalmente en pulmón, timo, hígado y riñón24.
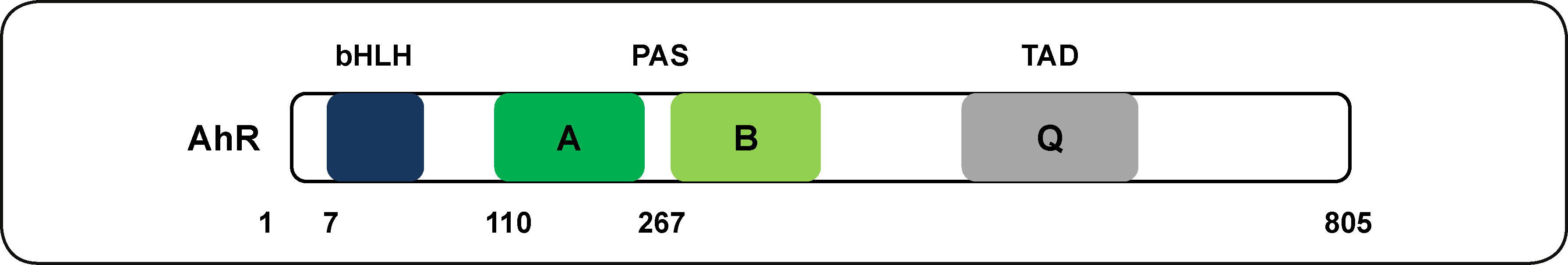
El AhR en mamíferos presenta tres dominios funcionales. Un dominio altamente conservado bHLH que consiste de 4 a 6 aminoácidos ácidos en el extremo amino terminal. Este dominio tiene la función de interacción con el ADN. El segundo dominio son dos repetidos de los dominios PAS (PAS A y PAS B) cada uno conteniendo aproximadamente 110 aminoácidos. Análisis estructurales, bioquímicos y de interacción de proteínas sugieren que el dominio PAS A está relacionado con la dimerización mientras que PAS B funciona como dominio de señalización6,25. El tercer domino (Q) es esencial en la transactivación, es un dominio poco conservado y se localiza en el extremo carboxilo terminal26 (Figura 2).
; Q, dominio rico en glutamina; TAD, dominio de activación transcripcional; LBD, dominio de unión a ligando. Los números representan los aminoácidos de la proteína. Modificado de Wu et al.6.")
Estructura del AhR de humano. Se muestran los dominios funcionales A y B, secuencias de repetidos de nucleótidos presentes en el dominio PAS; bHLH, dominio con estructura básica de hélice-bucle-hélice; dominio de unión de la proteína de choque térmico de 90 KDa (Hsp90); Q, dominio rico en glutamina; TAD, dominio de activación transcripcional; LBD, dominio de unión a ligando. Los números representan los aminoácidos de la proteína. Modificado de Wu et al.6.
La activación del AhR está dada por la unión de un ligando. Estos ligandos son principalmente xenobióticos, de naturaleza hidrofóbica y con estructura plana. Pueden ser de origen natural, como el caso de los metabolitos secundarios de plantas o producto de combustiones incompletas, como es el caso de los PAH. Pueden ser también de origen sintético como los hidrocarburos aromáticos halogenados (HAH) o el 2,3,7,8-tetraclorodibenzo-p-dioxina (TCDD). Además de unirse a xenobióticos, se ha reportado que el AhR es capaz de unir compuestos endógenos como la bilirrubina27, lipoxina A428 y derivados del triptófano29–31.
El AhR en estado latente (sin un ligando unido) se localiza predominantemente anclado en el citoplasma de la célula como parte de un complejo heterodimérico 9S. Este complejo está formado por un dímero de la proteína de choque térmico 90 (Hsp90), una proteína de choque térmico con un peso de 23 KDa (p23) y una proteína de interacción con AhR parecida a la inmunofilina (AIP), también conocida como XAP2 o Ara9. Adicionalmente, se ha descrito la presencia de otras proteínas citosólicas incluyendo la chaperona (Cdc37) y una cinasa de tirosina (c-src)34,35 (Figura 3).
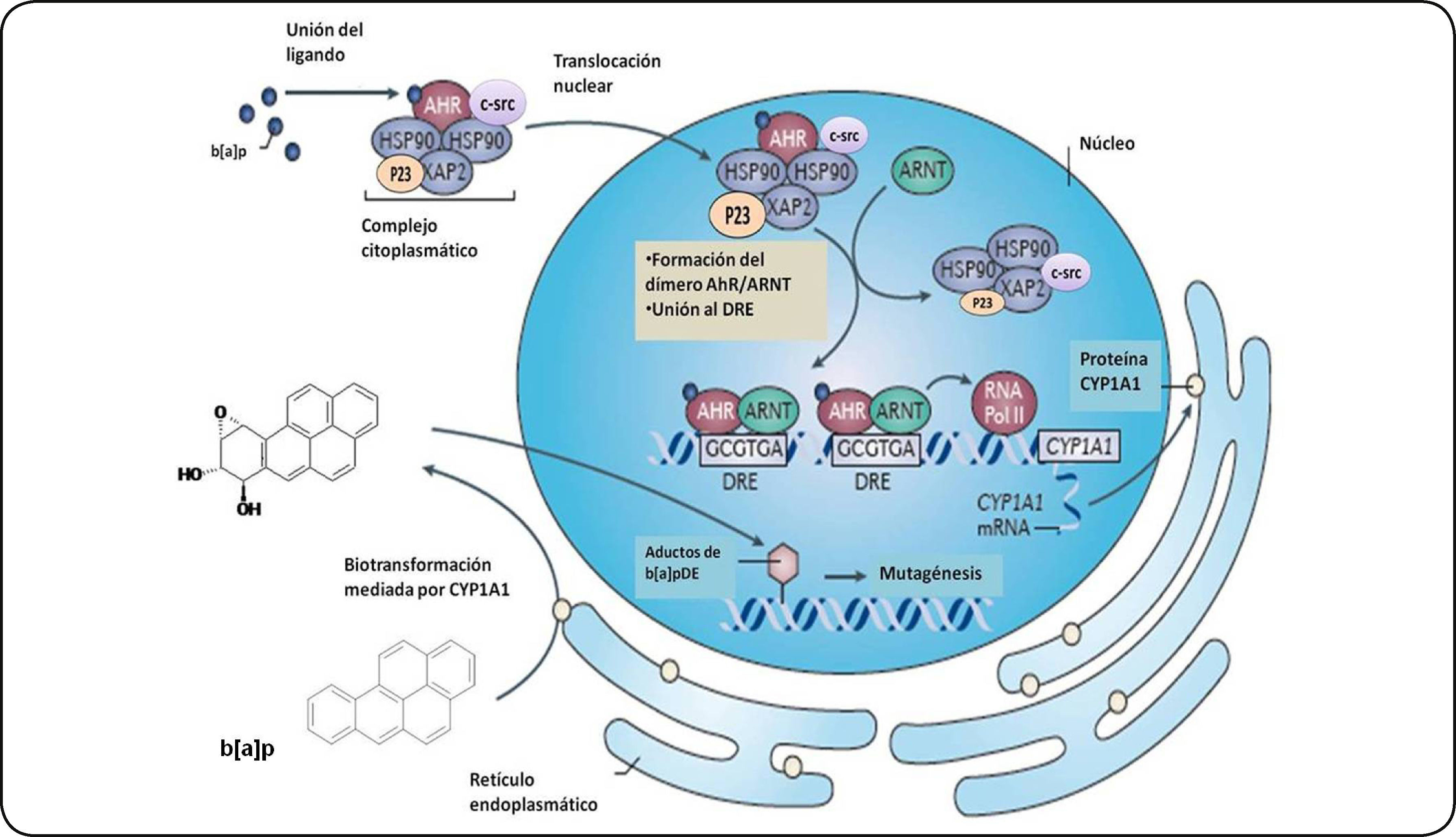
![Activación del AhR. El receptor en su forma inactiva (sin un ligando unido) se localiza en el citoplasma celular formando un complejo con un dímero de la proteína de choque térmico 90 (Hsp90), una proteína de choque térmico con un peso de 23 KDa (P23) y una proteína de interacción con el AhR parecida a la inmunofilina (AIP), también conocida como XAP2, y una cinasa de tirosina (c-src). Una vez unido su ligando, el complejo AhR se transloca al núcleo y forma un heterodímero con la proteina ARNT. Este heterodímero es capaz de unirse a los XRE (GCGTGA) y reclutar coactivadores y así favorecer la transcripción de sus genes blanco como el CYP1A1. Este citocromo metabolizará al b[a]p en intermediarios que pueden interactuar con el ADN para formar aductos iniciando así un proceso carcinogénico. Modificado de Murray et al.50.](https://static.elsevier.es/multimedia/1405888X/0000001900000001/v1_201602260016/S1405888X16000073/v1_201602260016/es/main.assets/gr3.jpeg?xkr=ue/ImdikoIMrsJoerZ+w997EogCnBdOOD93cPFbanNcNGSBFqzjIokZJFmIUKi41T4Z3ZEi/PmzFBpiTBVG8wrSKy0qRNQk3YX2SxjR9ufebUiBxCfBUuWB1SYJeC64CIhNhmWqo/phyea221AiD2Zmo5WO5TyK/wwQEDQdBzXpdhTGc4WRCpCSHYvnyNIG935SYIMA4/jSQPdOnGUvNUbUrml8Ka/cL/5bEAlKv2VIv3IYhywtrJQhfuaWcXLD91bGJIijTF4Hd4CY50f2E7Q== "Activación del AhR. El receptor en su forma inactiva (sin un ligando unido) se localiza en el citoplasma celular formando un complejo con un dímero de la proteína de choque térmico 90 (Hsp90), una proteína de choque térmico con un peso de 23 KDa (P23) y una proteína de interacción con el AhR parecida a la inmunofilina (AIP), también conocida como XAP2, y una cinasa de tirosina (c-src). Una vez unido su ligando, el complejo AhR se transloca al núcleo y forma un heterodímero con la proteina ARNT. Este heterodímero es capaz de unirse a los XRE (GCGTGA) y reclutar coactivadores y así favorecer la transcripción de sus genes blanco como el CYP1A1. Este citocromo metabolizará al b[a]p en intermediarios que pueden interactuar con el ADN para formar aductos iniciando así un proceso carcinogénico. Modificado de Murray et al.50.")
Activación del AhR. El receptor en su forma inactiva (sin un ligando unido) se localiza en el citoplasma celular formando un complejo con un dímero de la proteína de choque térmico 90 (Hsp90), una proteína de choque térmico con un peso de 23 KDa (P23) y una proteína de interacción con el AhR parecida a la inmunofilina (AIP), también conocida como XAP2, y una cinasa de tirosina (c-src). Una vez unido su ligando, el complejo AhR se transloca al núcleo y forma un heterodímero con la proteina ARNT. Este heterodímero es capaz de unirse a los XRE (GCGTGA) y reclutar coactivadores y así favorecer la transcripción de sus genes blanco como el CYP1A1. Este citocromo metabolizará al b[a]p en intermediarios que pueden interactuar con el ADN para formar aductos iniciando así un proceso carcinogénico. Modificado de Murray et al.50.
La activación del AhR se caracteriza por su translocación al núcleo y la disociación del complejo al cual está unido. Una vez en el núcleo, el AhR, formará un heterodímero con la proteína translocador nuclear del receptor de arilos (ARNT). Este heterodímero AhR/ARNT interaccionará con proteínas acetil transferasas de histonas y factores remodeladores de cromatina, lo que resulta en la unión del complejo AhR/ARNT a una secuencia de ADN consenso (GCGTGA) conocida como elemento de respuesta a xenobióticos o elementos de respuesta a dioxinas (XRE, DRE). Esta secuencia está localizada aproximadamente a 1 Kb río arriba de sus genes blanco36. Dentro de estos genes se incluyen aquéllos que codifican enzimas de la fase I del metabolismo de xenobióticos, representada por citocromos P450 (CYP450) y de la fase II la cual incluye a las glutatión S-transferasas (GST) y las UDP glucuronil transferasas (UGT). Los genes de la fase I, regulados por el AhR/ARNT, sólo incluyen a los miembros de la familia CYP1 de los CYP450 (CYP1A1, CYP1A2, CYP1B1)37,38. La expresión de otros CYP450, como CYP3 y CYP4, está regulada por los receptores nucleares CAR, PXR y PPAR los cuales dimerizan con el receptor RXR39.
De acuerdo con Hahn15, la habilidad del AhR de unir xenobióticos y mediar una respuesta adaptativa que involucra la inducción de enzimas metabolizadoras de xenobióticos, puede ser una función exclusiva de los vertebrados debido a que en los invertebrados el AhR no es capaz de unir xenobióticos. En estos últimos, el AhR participa en procesos de desarrollo, por ejemplo, en D. melanogaster es esencial para el correcto desarrollo de los segmentos distales de las antenas y las alas22,40 mientras que en C. elegans, el AhR participa en la diferenciación neuronal41,42.
REGULACIÓN DE LA VÍA DEL AHRDesde su descubrimiento en mamíferos se pensó que la única función del AhR era la de participar en la respuesta adaptativa al estrés celular, actuando como un sensor de señales endógenas o inducidas por xenobióticos. Sin embargo, existe evidencia experimental que vincula al AhR con la alteración de diversos procesos celulares como proliferación celular, apoptosis, diferenciación, promoción de tumores, reproducción y respuesta inmune43. Ésto sugiere que además de ser un sensor de xenobióticos, el AhR es esencial en la homeostasis celular. No obstante, los mecanismos por los cuales desempeña este último papel son poco conocidos. Por otro lado, la capacidad de respuesta del AhR difiere entre especies y tipo celular, lo que puede ser un indicativo de las posibles interacciones con diferentes vías de señalización y de la complejidad de su papel. Debido a que el AhR está involucrado en diversos procesos celulares, su señalización debe ser estrictamente regulada. Por este motivo, las células han desarrollado diferentes sistemas para evitar una excesiva señalización. Al ser una señal (ligando) el factor regulador de su transcripción, el mecanismo más directo de regular la vía es eliminar la señal. La activación del AhR conlleva a una sobreexpresión de enzimas de la fase I y II del metabolismo de xenobióticos las cuales oxidan y forman derivados de los ligandos, facilitando su remoción por medio de transportadores de membrana dependientes de ATP44. Otro mecanismo por el cual la célula es capaz de disminuir la señalización es por medio de la degradación vía proteosoma 26 S45–47. La actividad del AhR también puede ser modulada por la secuencia de exportación nuclear (NES) presente en el AhR. Esta secuencia permite que el AhR, en su forma activa, pueda ser exportado del núcleo hacia el citoplasma y de esa manera, evitar la inducción de la transcripción génica48. Por otro lado, la activación del AhR induce la transcripción del represor del AhR (AhRR). Sin embargo, el mecanismo exacto por el cual el AhRR reprime la vía del AhR no es conocida49, aunque se ha sugerido que el AhRR es capaz de competir directamente con el AhR in vitro, formando un dímero con el ARNT y, de esta manera, evitar la transcripción de sus genes blanco49.
BENZO[A]PIRENOUna de las principales moléculas capaces de activar al AhR e inducir su vía de señalización es el b[a]p. Este compuesto fue el primer carcinógeno detectado en el humo del tabaco y fue uno de los carcinógenos más estudiados durante el siglo pasado51 (Figura 4). El b[a]p es un hidrocarburo policíclico aromático formado por la combustión incompleta de materia orgánica, es un contaminante persistente y ubicuo en el ambiente. Las principales fuentes de exposición a b[a]p son las carnes cocinadas al carbón, la quema de combustibles fósiles, la quema de leña y el humo de tabaco52. Posterior a la exposición a b[a]p, éste puede distribuirse en distintos órganos como el hígado, riñón y sangre53–56. Por otro lado, la naturaleza lipofílica del b[a]p favorece su almacenamiento en tejidos grasos incluyendo glándulas mamarias y médula ósea. También se sabe que el b[a]p es capaz de cruzar la barrera hematoencefálica y la placenta57. A pesar de que el b[a]p puede metabolizarse en diversos órganos, se ha reportado que los metabolitos de b[a]p se concentran principalmente en el pulmón, específicamente en el tejido bronco epitelial58. Las células epiteliales del tracto respiratorio son las primeras en estar en contacto con las partículas de b[a]p presente en el ambiente. Se estima que una persona fumadora puede estar expuesta a concentraciones promedio de 20-40 ng de b[a]p por cigarro59.
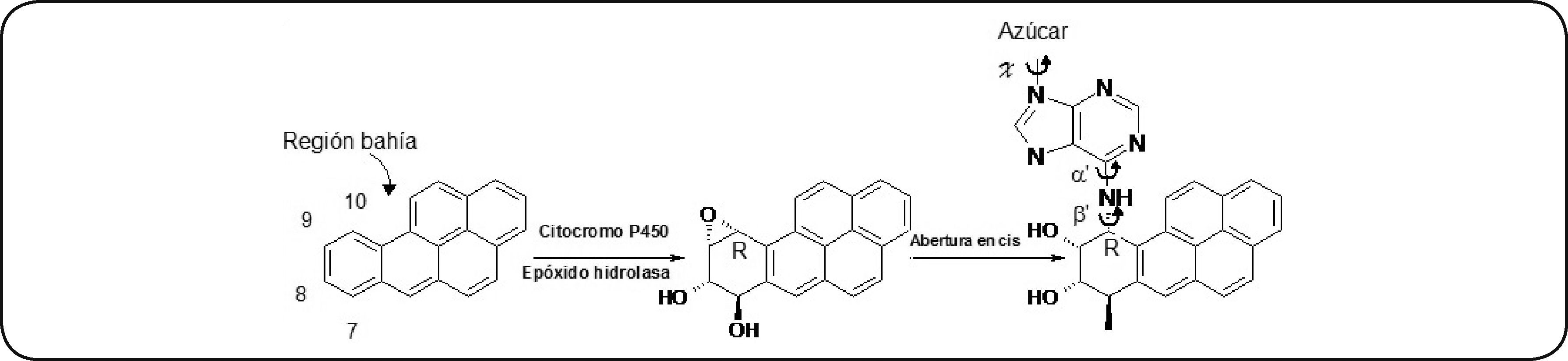
![Estructura química del b[a]p. Se representa la formación del metabolito benzo[a]pireno diol epóxido (b[a]pDE) mediada por CYP450 y epóxido hidrolasas, así como la formación de un aducto en el ADN dado por la apertura en cis del diol epóxido (DE) por el grupo amino del carbono 6 de la adenina. Modificado de Ling et al.63.](https://static.elsevier.es/multimedia/1405888X/0000001900000001/v1_201602260016/S1405888X16000073/v1_201602260016/es/main.assets/gr4.jpeg?xkr=ue/ImdikoIMrsJoerZ+w997EogCnBdOOD93cPFbanNcNGSBFqzjIokZJFmIUKi41T4Z3ZEi/PmzFBpiTBVG8wrSKy0qRNQk3YX2SxjR9ufebUiBxCfBUuWB1SYJeC64CIhNhmWqo/phyea221AiD2Zmo5WO5TyK/wwQEDQdBzXpdhTGc4WRCpCSHYvnyNIG935SYIMA4/jSQPdOnGUvNUbUrml8Ka/cL/5bEAlKv2VIv3IYhywtrJQhfuaWcXLD91bGJIijTF4Hd4CY50f2E7Q== "Estructura química del b[a]p. Se representa la formación del metabolito benzo[a]pireno diol epóxido (b[a]pDE) mediada por CYP450 y epóxido hidrolasas, así como la formación de un aducto en el ADN dado por la apertura en cis del diol epóxido (DE) por el grupo amino del carbono 6 de la adenina. Modificado de Ling et al.63.")
Estructura química del b[a]p. Se representa la formación del metabolito benzo[a]pireno diol epóxido (b[a]pDE) mediada por CYP450 y epóxido hidrolasas, así como la formación de un aducto en el ADN dado por la apertura en cis del diol epóxido (DE) por el grupo amino del carbono 6 de la adenina. Modificado de Ling et al.63.
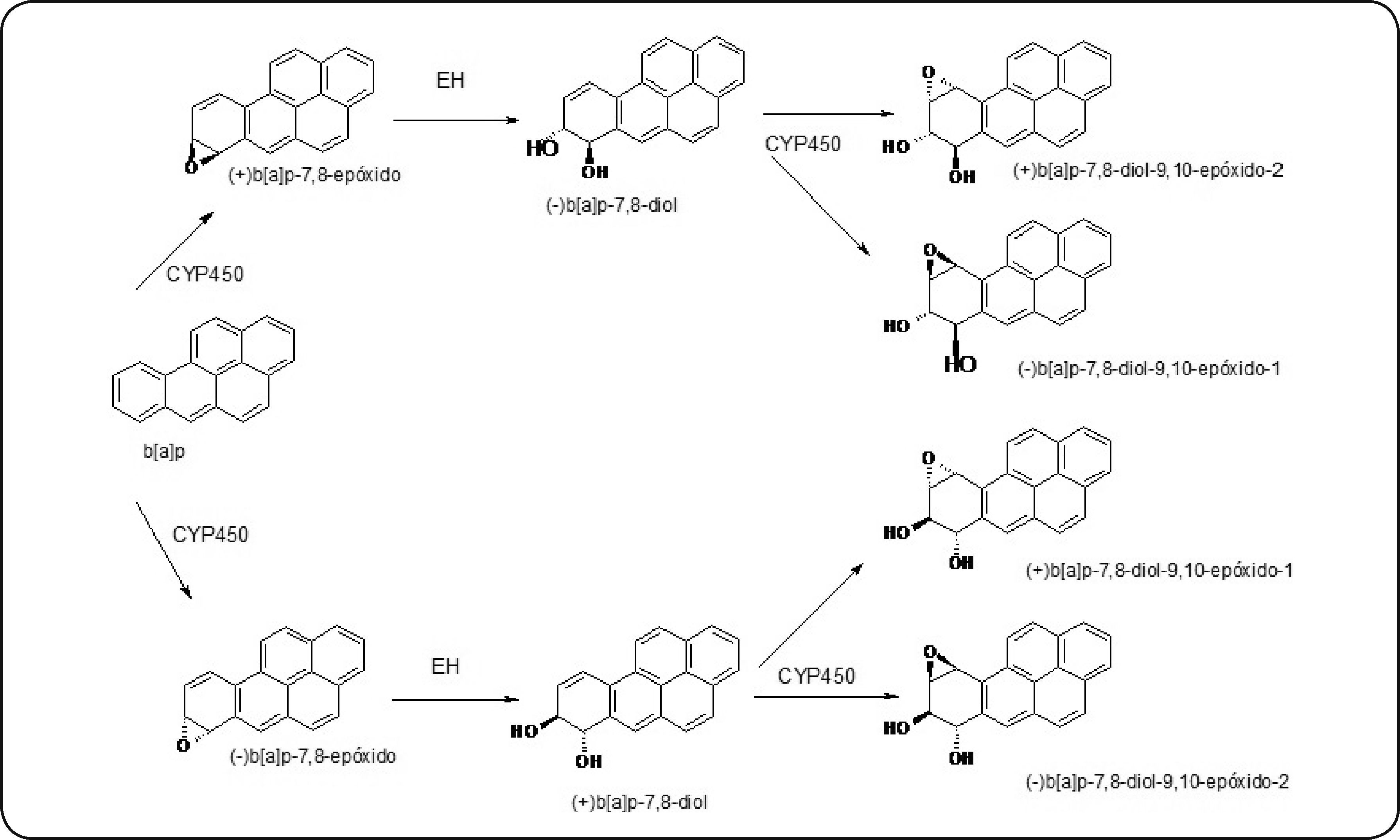
El metabolismo de b[a]p tiene especial importancia ya que aunque no es tóxico per se, su efecto carcinógeno reside en la formación de metabolitos primarios y secundarios potencialmente tóxicos que son capaces de interactuar con constituyentes celulares e inducir un daño celular. Esto mediante la vía de señalización del AhR60 la cual involucra la inducción de genes que codifican enzimas de las fases I y II del metabolismo de xenobióticos.
En la primera fase, el b[a]p es oxidado por el CYP1A1 y produce (+)- y (-)-b[a]p- 7, 8-óxidos, teniendo una tasa de conversión mucho más alta del enantiómero (+)-. Posteriormente, la epóxido hidrolasa (EH) hidroliza estos óxidos y se forman (-)- y (+)-b[a]p-7, 8-dioles, que son oxidados una vez más por CYP1A1 para formar (-)-b[a]p-7,8-diol-9, 10-epóxido-1, (+)-b[a]p-7,8-diol-9, 10-epóxido-2, (+)-b[a]p-7,8-diol-9, 10-epóxido-1 y (-)-b[a]p-7,8-diol-9, 10-epóxido-2 (b[a]pDE)61,62, como se muestra en la Figura 5.
![Vía metabólica mediante la cual el b[a]p se bioactiva a b[a]pDE, metabolitos altamente reactivos. Modificado de Shimada64.](https://static.elsevier.es/multimedia/1405888X/0000001900000001/v1_201602260016/S1405888X16000073/v1_201602260016/es/main.assets/gr5.jpeg?xkr=ue/ImdikoIMrsJoerZ+w997EogCnBdOOD93cPFbanNcNGSBFqzjIokZJFmIUKi41T4Z3ZEi/PmzFBpiTBVG8wrSKy0qRNQk3YX2SxjR9ufebUiBxCfBUuWB1SYJeC64CIhNhmWqo/phyea221AiD2Zmo5WO5TyK/wwQEDQdBzXpdhTGc4WRCpCSHYvnyNIG935SYIMA4/jSQPdOnGUvNUbUrml8Ka/cL/5bEAlKv2VIv3IYhywtrJQhfuaWcXLD91bGJIijTF4Hd4CY50f2E7Q== "Vía metabólica mediante la cual el b[a]p se bioactiva a b[a]pDE, metabolitos altamente reactivos. Modificado de Shimada64.")
Vía metabólica mediante la cual el b[a]p se bioactiva a b[a]pDE, metabolitos altamente reactivos. Modificado de Shimada64.
La fase II consiste en la conjugación de los b[a]pDE generados en la fase I con moléculas polares, y de esta manera evitar su interacción y un posible daño a constituyentes celulares como el ADN. Las enzimas de la fase II que catalizan la conjugación tienen diferente especificidad por sus sustratos. El nivel de expresión depende del tipo de tejido y estados de desarrollo del organismo. Por otro lado, pueden ser inducibles o inhibidas por xenobióticos65. Se han descrito diversas enzimas que participan en la fase II, por ejemplo, sulfotransferasas (SULT)66, UDP-glucuronosiltransferasas (UGT)67,68, DT-diaforasas o NAD(P)H:quinona oxidorreductasas (NQO), NAD(P)H:menadiona reductasas (NMO)69, epóxido hidrolasas (EH)70, glutatión S-transferasas (GST)71 y N-acetiltransferasas (NAT)72. Sin embargo, las GST son las principales enzimas que participan en esta fase73. El patrón de distribución y expresión de las GST es complejo, incluso, los diferentes tipos celulares presentes en un mismo tejido tienen distintos patrones de expresión. Así mismo, los patrones de distribución interindividual también pueden variar. Por ejemplo, en el humano los patrones de expresión de las GST en el pulmón pueden variar debido a la presencia de polimorfismos en genes que codifican a las enzimas74.
Las GST catalizan la conjugación de glutatión (GSH) con b[a]pDE, lo que generalmente incrementa la hidrofobicidad de b[a]pDE y por lo tanto aumenta su excreción por medio de la bilis u orina65. Este mecanismo es considerado como la vía de inactivación de b[a]pDE más importante75,76. Las tres isoformas de las GST mejor conocidas son las GST μ, θ y π; las isoformas μ y π tienen múltiples sustratos que incluyen al b[a]pDE, acroleína y otros carbonilos insaturados generados por peróxidos lipídicos y daño oxidante al ADN77–79. La isoforma de las GST μ presenta la mayor actividad enzimática en la conjugación de b[a]pDE seguida de las isoformas π y θ 80,81.
B[A]P COMO CARCINÓGENOComo se mencionó anteriormente, el b[a]p es un compuesto inerte. La capacidad mutagénica de este compuesto reside en su biotransformación y producción de metabolitos de b[a]p, principalmente b[a]pDE. Los b[a]pDE son considerados metabolitos carcinógenos debido a su capacidad para unirse covalentemente al ADN (formación de aductos) en los grupos amino de la posición C6 de la adenina o C2 de la guanina (Figura 4)82,83. Si estos aductos no son reparados pueden causar mutaciones en genes capaces de conducir a una transformación celular. Un ejemplo clásico de ello son los aductos de b[a]pDE que se han encontrado en sitios específicos dentro de la secuencia del gen TP5384. Los aductos de b[a]pDE en proteínas, como la hemoglobina, son considerados marcadores de exposición. Por otro lado, se ha reportado que la formación de aductos se correlaciona con aspectos como: el tabaquismo, sexo, presencia de polimorfismos en genes que codifican enzimas que participan en la reparación de daño al ADN, la elevada expresión de enzimas metabolizadoras de carcinógenos y el riesgo de cáncer de pulmón85,86.
La actividad carcinogénica y mutagénica del b[a]pDE se le atribuye a la presencia del epóxido en la posición C-10, también llamada región bahía (Figura 4)87. Por otro lado, también se pueden formar metabolitos fenólicos directamente por una hidroxilación en la posición C-3 y C-6 del b[a]p88. Los 6-OH-b[a]p son oxidados por peroxidasas o CYP450 para formar cationes tóxicos 6-oxi-b[a]p que posteriormente son oxidados en posición 1,6-3,6- ó 6,12- vía hidroquinonas o semiquinonas y, de esta manera, formar quinonas89,90. Estas quinonas no son mutagénicas, pero las especies reactivas generadas por sus intermediarios son responsables de sus efectos tóxicos91,92.
EL AHR COMO MEDIADOR EN PROCESOS FISIOLÓGICOSHay trabajos que se han enfocado en estudiar y establecer el papel del AhR como mediador de una respuesta adversa a contaminantes ambientales como el b[a]p. Sin embargo, el alto grado de conservación del AhR en muchas especies animales sugiere que éste podría tener funciones independientes a la del metabolismo de los xenobióticos15. De hecho, la participación del AhR en procesos del desarrollo se propuso por los patrones de expresión del AhR y del ARNT observados durante la embriogénesis del ratón93. Por otro lado, se ha relacionado al AhR con los procesos celulares de proliferación, apoptosis, diferenciación y promoción de tumores, además, de participar en procesos relacionados con los sistemas inmune y de reproducción. Estas funciones podrían estar relacionadas con la presencia de compuestos endógenos como la bilirrubina27, lipoxina A428, y derivados del triptófano29 que podrían funcionar como ligandos del AhR. Finalmente, los trabajos realizados con ratones carentes del AhR por tres grupos independientes94–96, apoyan la idea de la participación del AhR en la homeostasis y el desarrollo.
LOS RECEPTORES ERBBLos receptores ErbB son receptores presentes en la membrana celular que convierten señales extracelulares en una respuesta biológica97,98. El factor de crecimiento epidermal (EGF) y los factores de crecimiento similares al EGF son proteínas extracelulares que funcionan como ligandos de este tipo de receptores. Los motivos estructurales en común que caracterizan los receptores ErbB son: un dominio extracelular de unión a ligando, un dominio transmembranal hidrofóbico y un dominio citoplasmático con actividad de cinasa de tirosinas97. Uno de los receptores ErbB más estudiados es el receptor ErbB-1, también conocido como receptor del factor de crecimiento epidermal (EGFR). Este receptor es activado por la unión del EGF o factores de crecimiento relacionados, como el factor de crecimiento transformante α (TGF-α), anfiregulina (AREG), epiregulina (EREG) o el factor de crecimiento parecido al EGF de unión a heparina, entre otros99. La unión de estos ligandos permite la homo o heterodimerización de los receptores ErbB, lo que resulta en la estimulación de la actividad intrínseca de cinasa de tirosina, la cual consiste en la fosforilación de residuos de tirosina presentes en el dominio citoplasmático del receptor100. Estos dominios fosforilados funcionan como sitios de acoplamiento para moléculas de señalización similares a adaptadores los cuales activan directamente las principales vías de transducción de señales en la célula. Por lo tanto, traducen una señal determinada en una respuesta biológica. Subsecuentemente, las señales inducidas son atenuadas mediante la internalización de las moléculas de factores de crecimiento101. Esta vía de señalización está involucrada en la regulación de varios procesos celulares como la proliferación, diferenciación y migración. Su expresión es anormalmente elevada en la mayoría de los cánceres relacionados con células epiteliales102. La administración exógena de factores de crecimiento tiene efectos biológicos adversos como la síntesis de ácidos grasos103, inhibición de la fusión del paladar104 o la promoción de tumores en la piel105.
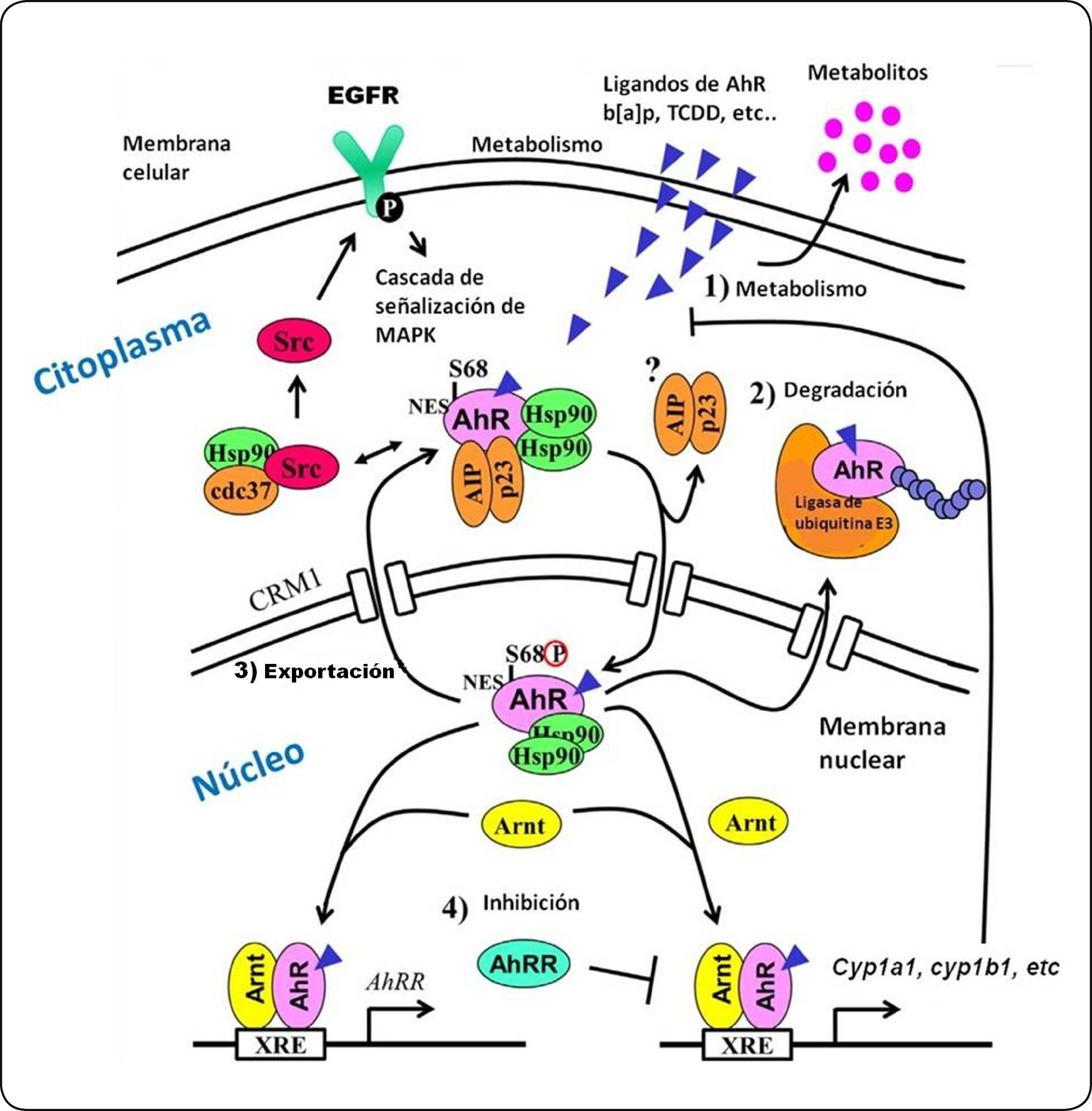
ACTIVACIÓN DE LA VÍA DEL AHR Y SU INTERCOMUNICACIÓN CON LA VÍA DE RECEPTORES TIPO ERBBExiste evidencia de que los efectos causados por TCDD son similares a los efectos causados por el EGF106–109. Esto sugiere que el TCDD (sin ser un ligando del EGFR) y el EGF afectan las mismas estructuras celulares. Los primeros indicios de la interferencia del TCDD con la vía de señalización del EGF son los resultados reportados por Madhukar et al.110 quienes compararon los efectos adversos de la administración del TCDD con el EGF en un modelo de ratón y observaron que animales expuestos al TCDD y al EGF tuvieron una apertura temprana de los párpados, erupción prematura de piezas dentales y reducción del peso corporal y del timo. A nivel celular, los autores observaron un aumento en la actividad de proteínas cinasas acompañado de una disminución en la capacidad del EGFR para unir el EGF marcado radioactivamente110. Estos resultados indicaron, por primera vez, que de alguna manera el TCDD es capaz de activar la vía de señalización mediada por el EGF, lo cual puede ser una explicación de los efectos pleiotrópicos causados por una exposición a este xenobiótico. Estudios posteriores in vivo e in vitro demostraron que la exposición al TCDD incrementa la actividad de proteínas cinasas, particularmente la proteína cinasa C (PKC) y proteínas cinasas de tirosinas111–113. Posteriormente, se demostró que los efectos mencionados también se pueden presentar en tejidos pancreáticos y adipocitos. Por lo tanto, los efectos no están restringidos a tejidos hepáticos114,115. Una explicación lógica de este fenómeno podría ser que el TCDD de alguna manera activa al EGFR de forma similar a como lo hace el EGF116,117. Sin embargo, el TCDD no es un ligando del EGFR. No obstante, la exposición al TCDD conlleva a alteraciones en los niveles del TGF-α, un ligando conocido del EGFR, como lo mencionan Choi et al118 y Gaido et al.119 donde demostraron que en queratinocitos humanos que fueron expuestos al TCDD se encontró un aumento en la expresión del TGF-α. En contraste con estos resultados, se ha reportado que el TCDD disminuye la expresión de TGF-α durante la formación del paladar (palatogénesis)120. Los experimentos realizados por Enan y Matsumura34 apoyan la idea de que debe existir una segunda vía de señalización dependiente del AhR distinta al modelo clásico, donde la activación del AhR conlleva la activación de genes blanco que cuentan con la secuencia consenso XRE. De esta forma, la identificación de la cinasa de tirosina c-src (pp60src) como parte del complejo citoplasmático asociado al AhR en su forma inactiva ha contribuido a esclarecer la relación entre la estimulación del EGFR y la vía del AhR y a generar un modelo hipotético por el que estas dos vías interaccionan entre sí35,121. La cinasa c-src en su forma soluble es capaz de activar al EGFR mediante la fosforilación de dos residuos específicos de tirosina, lo que resulta en la dimerización del receptor y en la activación de la cascada de señalización122,123. Como se ilustra en la figura 6, la unión de un ligando al AhR permite la disociación del complejo citoplasmático al que inicialmente se encuentra unido y la posterior liberación de c-src en el citoplasma121–124. La cinasa c-src se transloca a la membrana celular donde puede interactuar con el EGFR de forma bidireccional: la c-src puede unirse y fosforilar al EGFR induciendo así su activación125. La relevancia de c-src en la toxicidad mediada por el TCDD se descubrió usando ratones knock-out de c-src. Los ratones knock-out tratados con el TCDD mostraron una disminución de la actividad de la fosfoenolpiruvato carboxicinasa, de la acumulación de triglicéridos, de glicógeno, así como disminución en los efectos tóxicos en el hígado en comparación con los efectos encontrados en los ratones silvestres126–128.
 la biotransformación del ligando por medio de enzimas metabolizadoras de xenobióticos, (2) la degradación del AhR dependiente del proteosoma 26S, (3) la exportación del AhR del núcleo, y (4) la inducción de la proteína represora de los AhRR. Elementos que se muestran en la figura: AhRR, represor del receptor de hidrocarburos de arilo; AIP, proteína integradora del receptor de hidrocarburos de arilo; Arnt, translocador nuclear del receptor de hidrocarburos de arilo; Cdc37, Proteína del ciclo de división celular 37; Cyp1a1, citocromo P4501A1; Hsp90, proteína de choque térmico de 90 KDa; MAPK, proteínascinasas activadas por mitógenos; NES, señal de exportación nuclear; p23, proteína chaperona de Hsp90 de 23 KDa; S68, serina 68; Src, proteína cinasa de tirocinas Src; XRE, elemento de respuesta a xenobióticos. Modificado de Hao y Whitelaw139.")
El AhR y su intercomunicación con el EGFR. La interacción entre el AhR y su ligando permite su translocación al núcleo y la liberación de las proteínas a las cuales se encontraba asociado. Ya en el núcleo, el AhR dimeriza con la proteína del ARNT e induce la transcripción de genes blanco. Por su parte, la proteína c-src fosforila al EGFR, iniciando así la cascada de señalización mediada por MAPK. La actividad del AhR es regulada por: (1) la biotransformación del ligando por medio de enzimas metabolizadoras de xenobióticos, (2) la degradación del AhR dependiente del proteosoma 26S, (3) la exportación del AhR del núcleo, y (4) la inducción de la proteína represora de los AhRR. Elementos que se muestran en la figura: AhRR, represor del receptor de hidrocarburos de arilo; AIP, proteína integradora del receptor de hidrocarburos de arilo; Arnt, translocador nuclear del receptor de hidrocarburos de arilo; Cdc37, Proteína del ciclo de división celular 37; Cyp1a1, citocromo P4501A1; Hsp90, proteína de choque térmico de 90 KDa; MAPK, proteínascinasas activadas por mitógenos; NES, señal de exportación nuclear; p23, proteína chaperona de Hsp90 de 23 KDa; S68, serina 68; Src, proteína cinasa de tirocinas Src; XRE, elemento de respuesta a xenobióticos. Modificado de Hao y Whitelaw139.
La participación de la cinasa c-src en alteraciones celulares promovidas por una exposición al TCDD también se ha observado en líneas celulares. Por ejemplo, en la línea celular derivada de cáncer de mama (MCF10A) el TCDD fue capaz de actuar como antagonista en el crecimiento y la diferenciación celular inducidos por insulina. Mediante el uso de inhibidores farmacológicos, los autores muestran que las propiedades antagónicas del TCDD son mediadas por la activación de la cinasa c-src y de las cinasas extracelulares reguladas por señal 1 y 2 (ERK1/2)129. Por otro lado, Tannheimer et al.130 reportaron que los ligandos del AhR, b[a]p y el TCDD fueron capaces de actuar como agonistas en la vía de señalización del factor de crecimiento similar a la insulina en células MCF10A resultando en un aumento en la estimulación del crecimiento celular. Estos trabajos muestran que la exposición a determinados xenobióticos como b[a]p y TCDD induce la activación de las vías de señalización activadas por mitógenos (MAPK), algunas de las cuales forman parte de la vía de señalización del EGFR131–133. Por otro lado, se ha reportado que la activación del AhR, además de inducir la transcripción del principal gen en el metabolismo de b[a]p (CYP1A1), también es capaz de inducir la expresión de la ciclooxigenasa-2 (COX-2), la cual se sabe, es dependiente de la activación del EGFR y su vía de señalización es mediada por las MAPK134. Estos resultados fueron confirmados usando ratones knock-out para el gen del AhR donde se muestra una disminución en la inducción de los mRNA de COX-2 y CYP1A1 posterior a un estrés inducido por radiación UV-B. Estos datos sugieren que el AhR, CYP1A1 y COX-2, están involucrados en procesos carcinogénicos en la piel134–136. Otro equipo de investigación reportó que en macrófagos humanos (THP-1) expuestos al TCDD se induce una sobreexpresión del TNF-α137. Los autores argumentan que este incremento se debe a la activación del EGFR y su vía de señalización mediada por ERK 1/2. De manera interesante, en el mismo trabajo fue posible bloquear la expresión del TNF-α con el compuesto a naftoflavona (α-NF), un antagonista del AhR. Sin embargo, no se logró este mismo resultado usando un inhibidor conocido de la cinasa c-src (PP2). Por lo tanto, este resultado sugiere que posiblemente exista otro mecanismo celular que permita la interconexión de la vía de señalización del AhR con la cascada de señalización del EGFR. Un posible mecanismo puede ser la vía de señalización de la proteína cinasa A (PKA) la cual se sabe, es capaz de regular la transcripción de ampiregulina (AREG) que es un ligando del EGFR138. Además, la exposición de células epiteliales orales a extractos de humo de tabaco induce la señalización mediada por cAMP/PKA y la activación de las proteínas de unión a los elementos de respuesta a cAMP (CREB), resultando en un incremento de los mRNA de AREG140. Interesantemente, en células pre tratadas con α-NF, la inducción de AREG promovida por el humo de tabaco es nula.
El hecho de que PKA promueve la activación de la vía del EGFR podría significar una conexión independiente de la cinasa c-src entre de las vías de AhR y el EGFR141. En líneas celulares de ratón y otros murinos, el TCDD induce la expresión de los ligandos del EGFR, AREG y epiregulina (EREG) de una forma dependiente del AhR/XRE142,143. Patel y su equipo de trabajo identificaron un XRE funcional en el promotor de la EREG en murinos, el cual no tiene homología con el gen en los humanos. Sin embargo, y con base en estos datos, se podría especular que la sobreexpresión de los ligandos del EGFR como la EREG y la AREG inducidos por el TCDD podría causar una activación autocrina retrasada del EGFR, que podría explicar la activación del EGFR mediada por el AhR en presencia del inhibidor de c-src. Por lo tanto, la conexión entre estas tres vías de señalización (AhR, c-src y el EGFR), puede representar un complejo de señalización por el cual compuestos como el b[a]p podrían ser capaces de interactuar con receptores tipo ErbB y, de esta manera desencadenar alteraciones en procesos celulares relacionados con el cáncer.
CONCLUSIONESLos trabajos reportados sobre la interacción del AhR con la vía del EGFR muestran la importancia de esta intercomunicación en procesos de estrés celular mediados por la exposición a xenobióticos. Adicionalmente, la sobreexpresión del AhR y del EGFR presente en algunos tumores como los de mama y de pulmón hace evidente la importancia de conocer los detalles de los mecanismos moleculares con miras al desarrollo de nuevas estrategias preventivas y terapéuticas contra enfermedades como el cáncer. Adicionalmente, se debe enfatizar que la mayoría de los estudios se han realizado con líneas celulares derivadas del cáncer o líneas celulares que no corresponden a los tejidos blanco de los compuestos estudiados o de sus metabolitos. Esto implica que en los modelos celulares derivados del cáncer existen alteraciones en la regulación de su ciclo celular y no poseen los elementos celulares necesarios para un correcto metabolismo de los compuestos estudiados. Para una mejor comprensión de las vías de señalización involucradas en procesos de biotransformación y su posible relación con procesos carcinogénicos, los trabajos deben ser realizados en células no transformadas, que cuenten con un sistema de biotransformación funcional y que el modelo corresponda al órgano blanco del compuesto estudiado.
AGRADECIMIENTOSAl Programa de Doctorado en Ciencias Biomédicas de la Universidad Nacional Autónoma de México. Al Consejo nacional de Ciencia y Tecnología (CONACyT) por la beca otorgada para los estudios de doctorado de Gerardo Vázquez Gómez (No. de becario 233838).