




La F1FO-ATP sintasa es un complejo enzimático que se encuentra ampliamente distribuido en las membranas transductoras de energía. Todas las ATPasas, incluidas las mitocondriales, cloroplastídicas y bacterianas, comparten similitudes estructurales y funcionales. Sin embargo, hay diferencias en su composición que dependen de la especie, siendo más compleja en organismos como Saccharomyces cerevisiae o Bos taurus. Es por ello que una mejor comprensión de la estructura de la F1FO-ATP sintasa contribuirá a un mayor conocimiento a nivel molecular tanto de la función como de la regulación de este complejo enzimático. En la actualidad, se sabe muy poco acerca de la organización estructural de las subunidades de la región FO. Considerando lo anterior, en este trabajo se presenta información concerniente a las proteínas intrínsecas del dominio FO de las ATP sintasas más investigadas a la fecha, así como de algunas otras subunidades de membrana que se encuentran presentes en organismos menos estudiados.
The F1FO-ATP synthase is a complex widely distributed in energy-transducing membranes. All ATPases, including the mitochondrial, chloroplastic and bacterial, share structural and functional similarities. However, there are differences in their composition that depend on the species, being more complex in organisms such as Saccharomyces cerevisiae or Bos taurus. This is why, a better understanding of the F1FO-ATP synthase structure will contribute to a greater knowledge at a molecular level, both of the function, and the regulation of this enzymatic complex. At present, very little is known about the structural organization of the subunits from the FO domain. Considering the former, this paper presents information concerning the intrinsic membrane proteins from the most researched F1FO-ATP synthases to date, as well as some other membrane subunits present in less studied organisms.
La F1FO-ATP sintasa, F1FO-ATPasa o complejo V (EC 3.6.3.14) produce la mayoría del ATP celular en los eucariontes y en las bacterias. Este complejo enzimático se encuentra en las membranas transductoras de energía, como la membrana interna mitocondrial, la membrana tilacoidal del cloroplasto, y la membrana plasmática bacteriana (Domínguez-Ramírez & Tuena de Gómez-Poyou, 2003) (Figura 1). En eucariontes, la ATP sintasa utiliza el potencial electroquímico generado por los complejos de la cadena respiratoria o fotosintética para sintetizar ATP (Leyva et al., 2003). En bacterias, esta enzima puede aprovechar como fuerza impulsora tanto el gradiente de protones como el de iones sodio, como en Propionigenium modestum y Acetobacterium woodii (Deckers-Hebestreit & Altendorf, 1996).
 del dominio F1. Una de las tres subunidades α ha sido removida para exponer a la subunidad γ, cabe resaltar que ésta abarca desde el eje central del dominio catalítico hasta el anillo de subunidad c y la subunidad a. De igual forma, el brazo periférico de cada modelo se muestra a la derecha. En algunas bacterias está compuesto por la subunidad δ y el dímero de subunidad b. Mientras que en otras especies bacterianas, así como en la enzima cloroplastídica, las dos subunidades b son copias homólogas, pero no idénticas (subunidad b y b’). En la enzima mitocondrial, el brazo periférico lo componen una copia de las subunidades b, d y F6. El dominio membranal de la enzima mitocondrial contiene subunidades con una hélice transmembranal (e, f, g, 8 (A6L), entre otras descritas en la Tabla I), las cuales no se encuentran en las ATPasas bacterianas y cloroplastídicas. Modificado de Walker, 2013.")
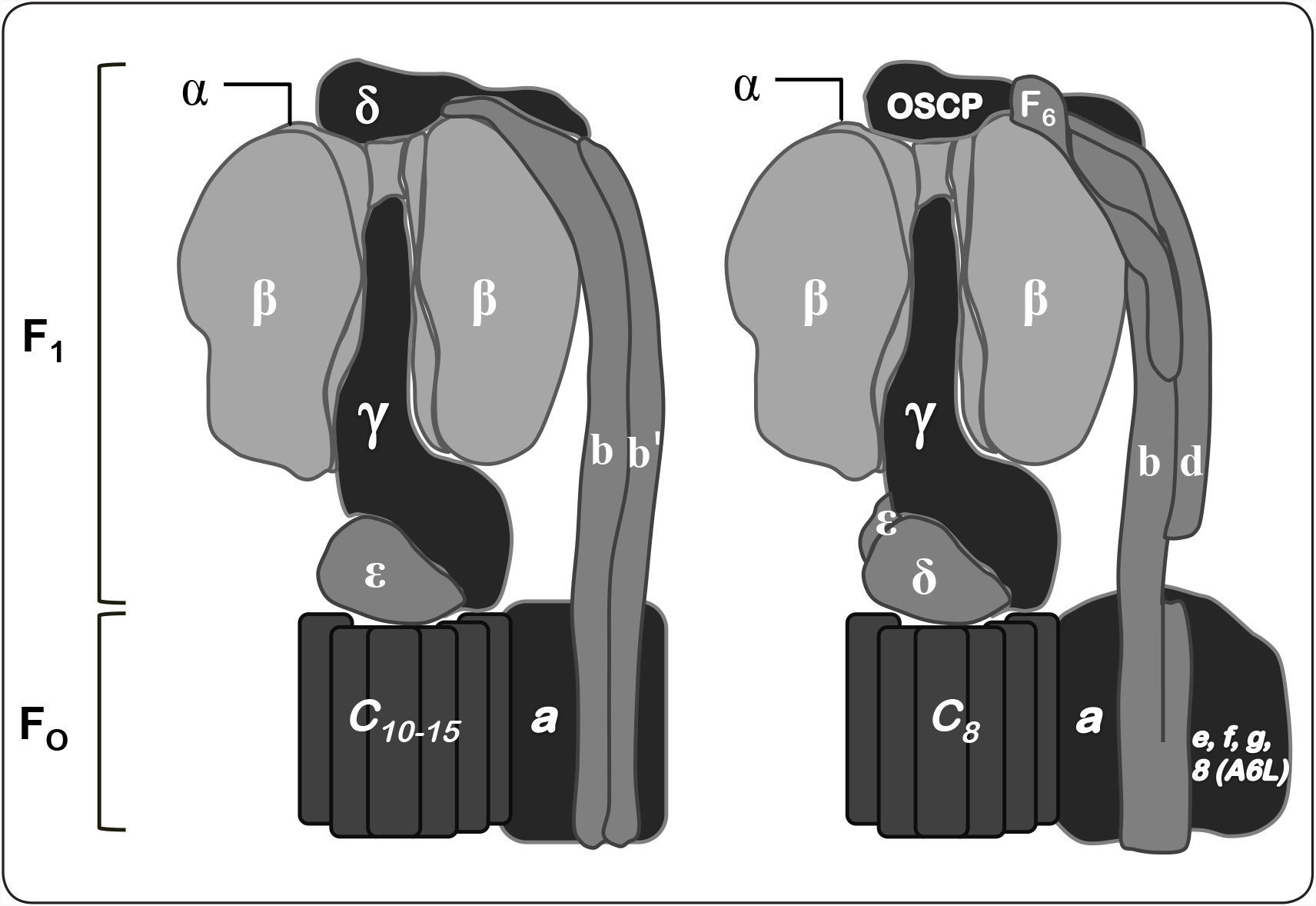
Organización polipeptídica de las F1FO-ATP sintasas. Las F-ATPasas bacterianas y cloroplastídicas se muestran a la izquierda, mientras que la mitocondrial se encuentra a la derecha. La parte superior de cada modelo contiene a las subunidades catalíticas (α3β3) del dominio F1. Una de las tres subunidades α ha sido removida para exponer a la subunidad γ, cabe resaltar que ésta abarca desde el eje central del dominio catalítico hasta el anillo de subunidad c y la subunidad a. De igual forma, el brazo periférico de cada modelo se muestra a la derecha. En algunas bacterias está compuesto por la subunidad δ y el dímero de subunidad b. Mientras que en otras especies bacterianas, así como en la enzima cloroplastídica, las dos subunidades b son copias homólogas, pero no idénticas (subunidad b y b’). En la enzima mitocondrial, el brazo periférico lo componen una copia de las subunidades b, d y F6. El dominio membranal de la enzima mitocondrial contiene subunidades con una hélice transmembranal (e, f, g, 8 (A6L), entre otras descritas en la Tabla I), las cuales no se encuentran en las ATPasas bacterianas y cloroplastídicas. Modificado de Walker, 2013.
Por otra parte, según las condiciones de la fuerza protón-motriz, la F1FO puede funcionar como ATP sintasa (síntesis de ATP) o ATPasa (hidrólisis de ATP), dicha regulación está determinada por la disponibilidad de los sustratos ADP y Pi, así como por el potencial electroquímico (Deckers-Hebestreit & Altendorf, 1996). Otro regulador importante de la ATP sintasa mitocondrial, exclusivo de eucariontes, es la proteína inhibidora (IF1). En bovino, la IF1 es una proteína básica de 84 aminoácidos capaz de inhibir la actividad del complejo enzimático en condiciones que favorecerían la hidrólisis de ATP, previniendo así el abatimiento de la función mitocondrial en ausencia del potencial electroquímico (Domínguez-Ramírez & Tuena de Gómez-Poyou, 2003). En este sentido, la ATP sintasa bacteriana posee un mecanismo similar, a través de un cambio conformacional de la subunidad ¿, que impide la rotación del complejo enzimático y de esta forma auto inhibe la rotación de la enzima en el sentido de la hidrólisis de ATP (García-Trejo et al., 2012).
Las F1FO-ATPasas, incluidas las mitocondriales, las cloroplastídicas y las bacterianas, se organizan en dos regiones estructural y funcionalmente distintas: a) un dominio membranal (FO), que participa en la translocación de iones (protones o sodio) y está compuesto básicamente por dos subunidades (a y c); y b) un dominio extrínseco de membrana (F1), que contiene los sitios catalíticos (subunidades α y β) (Deckers-Hebestreit & Altendorf, 1992); estos dominios se encuentran unidos a través de un tallo central (subunidades γ y ¿) y un brazo periférico (subunidad b en la ATPasa de Escherichia coli, subunidad b y b’ en las bacterias fotosintéticas, así como en la del cloroplasto; subunidad b, d, y F6 en la enzima mitocondrial; subunidad δ en la ATP sintasa bacteriana y cloroplastídica; subunidad OSCP en la ATPasa mitocondrial, mientras que el resto de las subunidades varía de acuerdo a la especie) (Figura 1) (Soubannier et al., 1999; Walker, 2013; Claggett et al., 2007).
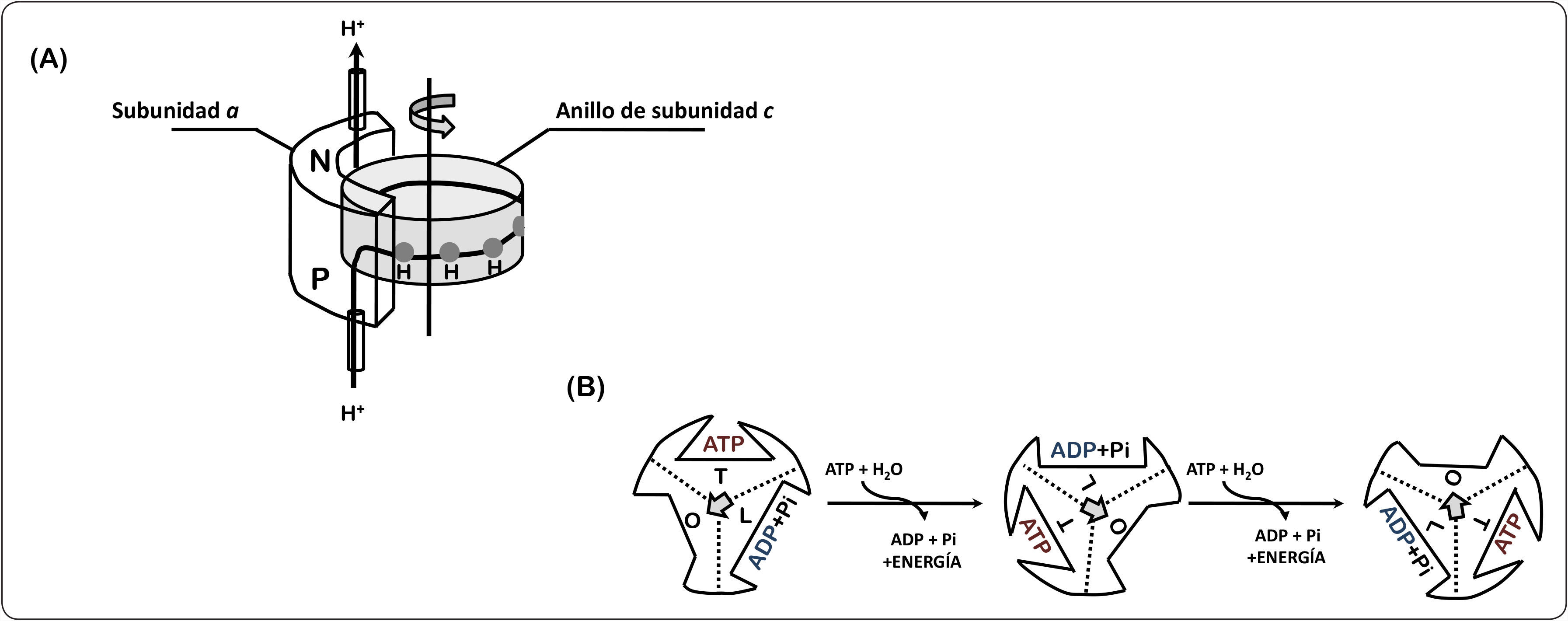
En cuanto a la síntesis de ATP, estudios funcionales y estructurales han mostrado que el canal de protones FO y la parte catalítica F1 se acoplan estructural y funcionalmente, en donde los protones atraviesan la membrana por un hemicanal formado entre las subunidades a y la subunidad c, los residuos funcionalmente importantes son la arginina o glutamina 239 de la subunidad a y el aspártico o glutámico 61 del anillo de subunidades de c, provocando el giro del anillo de proteolípidos formado por la subunidad c (Figura 2). Esta rotación hace girar al tallo central (subunidades γ y ¿) en movimientos sucesivos de 120°, provocando cambios conformacionales alternados consecutivos en las subunidades catalíticas (subunidades α y β) e induciendo la unión de los substratos (ADP y Pi), así como la síntesis y la liberación del ATP de acuerdo al mecanismo de cambio de unión propuesto por Boyer (Itoh et al., 2004).
 Los protones atraviesan la membrana del lado positivo (P) hacia el lado negativo (N) a favor del gradiente electroquímico a través de un hemicanal formado entre las subunidades a y el anillo de subunidades c generando una rotación en la dirección indicada (flecha). Cada subunidad c contiene un residuo de aspártico o glutámico (círculo gris), el cual es importante para la translocación de iones. (B) Las tres subunidades catalíticas beta adquieren diferentes estados conformacionales durante la síntesis de ATP: abierta (O), semiabierta (L) y cerrada (T). Cuando la subunidad pasa del estado cerrado al abierto se libera una molécula de ATP y a su vez se capta ADP y fosfato. Con la unión de los sustratos, la subunidad cambia a una conformación semiabierta, posteriormente en este sitio se lleva a cabo la reacción de formación del ATP y uno de los sitios catalíticos se abre para liberar el producto. Este ciclo se repite alternadamente en cada una de las subunidades beta que componen el dominio catalítico de la enzima. Modificado de Walker, 2013; Stewart et al., 2013.")
Mecanismo de la síntesis de ATP. (A) Los protones atraviesan la membrana del lado positivo (P) hacia el lado negativo (N) a favor del gradiente electroquímico a través de un hemicanal formado entre las subunidades a y el anillo de subunidades c generando una rotación en la dirección indicada (flecha). Cada subunidad c contiene un residuo de aspártico o glutámico (círculo gris), el cual es importante para la translocación de iones. (B) Las tres subunidades catalíticas beta adquieren diferentes estados conformacionales durante la síntesis de ATP: abierta (O), semiabierta (L) y cerrada (T). Cuando la subunidad pasa del estado cerrado al abierto se libera una molécula de ATP y a su vez se capta ADP y fosfato. Con la unión de los sustratos, la subunidad cambia a una conformación semiabierta, posteriormente en este sitio se lleva a cabo la reacción de formación del ATP y uno de los sitios catalíticos se abre para liberar el producto. Este ciclo se repite alternadamente en cada una de las subunidades beta que componen el dominio catalítico de la enzima. Modificado de Walker, 2013; Stewart et al., 2013.
El complejo enzimático de E. coli, la ATP sintasa estruc-turalmente más sencilla que se conoce, contiene ocho subunidades: las subunidades α3, β3, γ1, δ1 y ¿1 que constituyen el dominio F1, mientras que las subunidades a1, b2 y c10-12 conforman el dominio FO (Foster & Fillingame, 1982). En contraste al número de proteínas que constituyen a la ATP sintasa bacteriana, la composición polipeptídica de la enzima de otros organismos, como Saccharomyces cerevisiae o Bos taurus, es más compleja, presentando por lo menos 13 y 16 subunidades respectivamente (Tabla I) (Walker, 2013). En lo que respecta a la composición del brazo periférico, la ATPasa mitocondrial está compuesta por cuatro subunidades: b, d, F6 y OSCP (homólogo a la subunidad δ bacteriana). En el caso de la levadura, una sola subunidad b interactúa con las subunidades: 4, 8, d, f y h. Adicionalmente, en el dominio membranal de la enzima de bovino y en el de la levadura se encuentran las proteínas: e, g, i (conocida también como j y k), las cuales se han llamado supernumerarias, debido a que parecen no estar involucradas directamente en la síntesis de ATP, ya que no están presentes en la enzima bacteriana (Figura 1) (Fronzes et al., 2006).
Proteínas y subunidades asociadas a la F1FO-ATP sintasa de bacteria, levadura y bovino
| Subunidad | E. coli(1, 2) | S. cerevisiae(3) | B. taurus(3) | Sector |
|---|---|---|---|---|
| α | atpA (55.4) | ATP1 (54.9) | ATP5A1 (55.2) | F1 |
| β | atpD (50.4) | ATP2 (51.1) | ATP5B (51.7) | |
| γ | atpG (31.5) | ATP3 (30.6) | ATP5C1 (30.2) | |
| δ | atpH (19.3) | ATP16 (14.5) | ATP5D (15) | |
| ¿ | atpC (14.3) | ATP15 (6.6) | ATP5E (5.6) | |
| OSCP | ATP5 (20.8) | ATP5O (20.9) | ||
| IF1 | INH1 (7.3) | ATPIF1 (9.5) | ||
| a | atpB (30.1) | ATP6 (27.8) | MT-ATP6 (24.7) | FO |
| b | atpF (17.2) | ATP4 (23.2) | ATP5F1 (24.6) | |
| c | atpE (8.2) | ATP9 (7.7) | ATP5G1-3 (7.6) | |
| d | ATP7 (19.6) | ATP5H (18.5) | ||
| e | ATP21 (10.7) | ATP5I (8.1) | ||
| f | ATP17 (10.5) | ATP5J2 (10.1) | ||
| g | ATP20 (12.9) | ATP5L (11.2) | ||
| h | ATP14 (10.4) | |||
| i (j) | ATP18 (6.6) | |||
| k | ATP19 (7.5) | |||
| F6 | ATP5J (8.9) | |||
| 8 (A6L) | ATP8 (5.8) | MT-ATP8 (7.9) | ||
| AGP o DAPIT | USMG5 (6.3) | |||
| Proteína MLQ | MP68 (6.8) | |||
| Factor B | ATP5S (20.3) | |||
| Stf1 | STF1 (7.2) | |||
| Stf 2 | STF2 (9.4) | |||
| Sf l2 | TMA10 (9.7) |
En la Tabla se describe la composición polipeptídica y la distribución de las subunidades de la ATP sintasa de diferentes organismos, en donde se indica su nomenclatura y el gen que la codifica, así como su peso molecular estimado (kDa). 1Foster & Fillingame, 1982; 2Godinot & Di Pietro, 1986; 3Wittig & Schägger, 2008.
Estudios sobre la resolución de la estructura cristalina de la F-ATPasa intacta, se han frenado por la tendencia del complejo enzimático a disociarse cuando se extrae de la membrana. Sin embargo, un número de modelos atómicos se han obtenido para diversas regiones de la enzima de bovino y levadura (Jiko et al., 2015), incluyendo el dominio F1 (Abrahams et al., 1994), el subcomplejo F1-anillo de subunidad c (Stock et al., 1999), y la región del brazo periférico (Dickson et al., 2006; Rees et al., 2009). También hay información estructural de las enzimas de E. coli (Cingolani & Duncan, 2011; Roy et al., 2012), Caldalkalibacillus thermarum (Stocker et al., 2007), Geobacillus stearothermophilus (anteriormente Bacillus PS3) (Shirakihara et al., 2015) y de la α-proteobacteria Paracoccus denitrificans (Figuras 3 y 4) (Morales-Ríos et al., 2015).
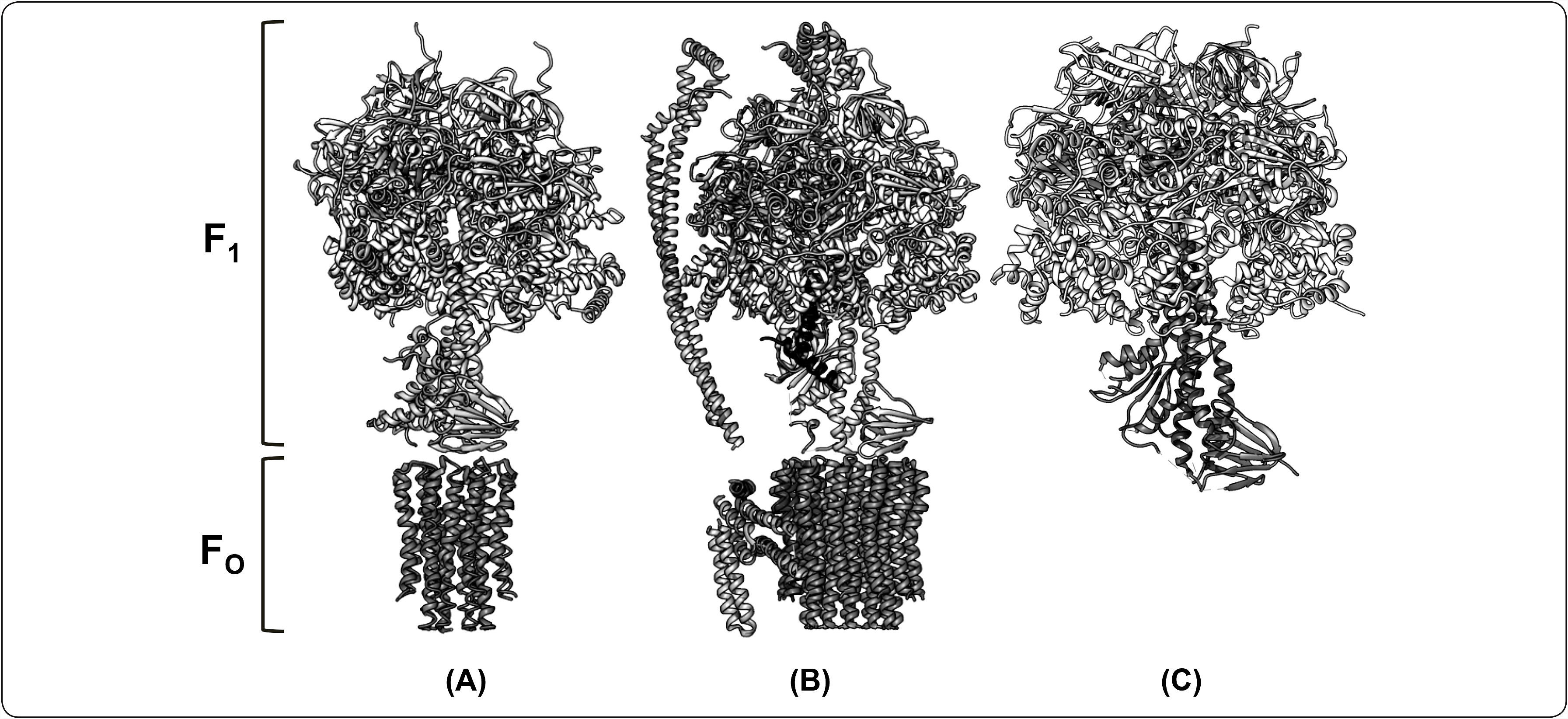
 Estructura cristalográfica del subcomplejo F1-subunidad c8 de la ATPasa de bovino (3.5Å de resolución, No. PDB 2XND). (Modificado de Rees et al., 2009). (B) Estructura de la ATP sintasa de P. denitrificans obtenida por difracción de rayos X (3.98Å de resolución, No. PDB 5DN6).(Morales-Ríos et al., 2015). (C) Estructura cristalográfica de la F1-ATPasa de Bacillus PS3 (TF1) (3.9Å de resolución, No. PDB 4XD7). (Modificada de Shirakihara et al., 2015). Cada uno de los modelos comprende a las subunidades del dominio catalítico (α3, β3) y el rotor central (γ, ¿). Adicionalmente, en el modelo estructural de (A) se representa a la subunidad δ y el anillo de subunidades c. Mientras que en (B), la estructura cristalográfica comprende el brazo periférico (δ, b,b’), el inhibidor (ζ) y el anillo de subunidades c.")
Comparación estructural de la ATP sintasa de diferentes organismos. A la izquierda de la figura se indica la región correspondiente al dominio F1 y FO para cada modelo presentado como una estructura de tipo listón. (A) Estructura cristalográfica del subcomplejo F1-subunidad c8 de la ATPasa de bovino (3.5Å de resolución, No. PDB 2XND). (Modificado de Rees et al., 2009). (B) Estructura de la ATP sintasa de P. denitrificans obtenida por difracción de rayos X (3.98Å de resolución, No. PDB 5DN6).(Morales-Ríos et al., 2015). (C) Estructura cristalográfica de la F1-ATPasa de Bacillus PS3 (TF1) (3.9Å de resolución, No. PDB 4XD7). (Modificada de Shirakihara et al., 2015). Cada uno de los modelos comprende a las subunidades del dominio catalítico (α3, β3) y el rotor central (γ, ¿). Adicionalmente, en el modelo estructural de (A) se representa a la subunidad δ y el anillo de subunidades c. Mientras que en (B), la estructura cristalográfica comprende el brazo periférico (δ, b,b’), el inhibidor (ζ) y el anillo de subunidades c.
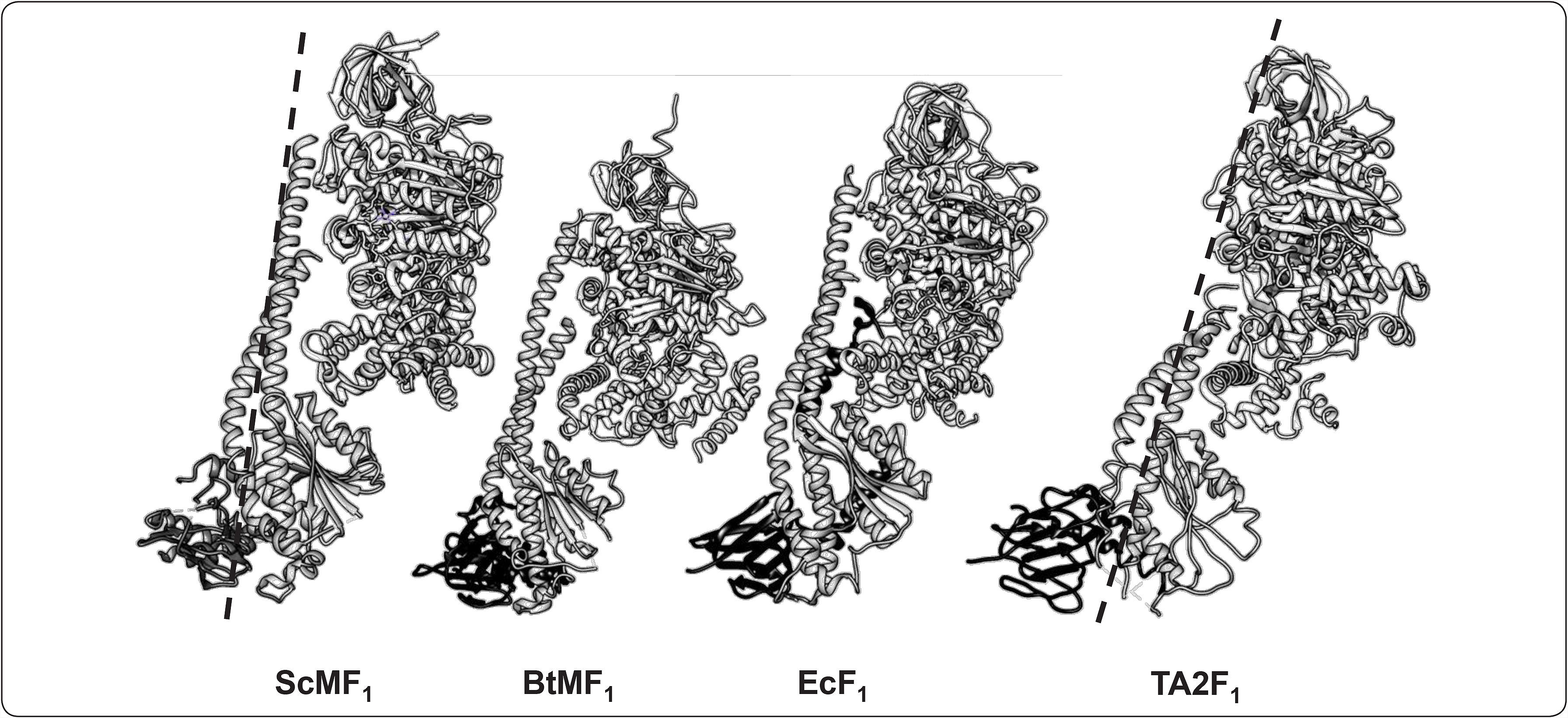
 y el sub-complejo F1-c8 de B. taurus (BtMF1 3.5Å, No. PDB 2XND); así como del sector F1 de la ATPasa de las especies bacterianas E. coli (EcF1 3.26Å, No. PDB 3OAA) y Bacillus sp. TA2.A1 (TA2F1 3Å, No. PDB 2QE7). En cada modelo se muestra a las subunidades α, β, γ y ¿. Asimismo, se compara el grado de torsión (línea punteada) del ScMF1 con respecto al de la bacteria termófila (TA2F1). Modificado de Stocker et al., 2007.")
Comparación estructural de la F1-ATPasa de diferentes organismos. Modelos tridimensionales atómicos representados como diagramas de listón. Estructuras cristalográficas de los sub-complejos F1-c10 de la ATP sintasa de S. cerevisiae (ScMF1 6.5Å, No. PDB 3ZYR) y el sub-complejo F1-c8 de B. taurus (BtMF1 3.5Å, No. PDB 2XND); así como del sector F1 de la ATPasa de las especies bacterianas E. coli (EcF1 3.26Å, No. PDB 3OAA) y Bacillus sp. TA2.A1 (TA2F1 3Å, No. PDB 2QE7). En cada modelo se muestra a las subunidades α, β, γ y ¿. Asimismo, se compara el grado de torsión (línea punteada) del ScMF1 con respecto al de la bacteria termófila (TA2F1). Modificado de Stocker et al., 2007.
En la actualidad, se sabe muy poco acerca de la organización estructural de las subunidades de la región FO. Hasta ahora, no se ha purificado a homogeneidad el dominio membranal de la ATPasa, ni su composición polipeptídica y tampoco establecido con certeza (Collinson et al., 1994). Considerando lo anterior, en este trabajo se presenta información concerniente a las proteínas intrínsecas del dominio FO de las ATP sintasas más investigadas a la fecha, así como de algunas otras subunidades de membrana que se encuentran presentes en organismos menos estudiados.
Dominio membranal de la F1FO-ATP sintasaComo se mencionó, en contraste con el detallado modelo estructural para el dominio F1 de la ATPasa de diferentes especies, la composición de subunidades y la información estructural del sector FO no es claro, en particular para la enzima mitocondrial. No obstante, todas las especies estudiadas a la fecha presentan tres subunidades comunes conocidas como a, b y c (Stock et al., 1999). Por este motivo, en principio se describirá la información estructural de estas subunidades, y posteriormente se abordarán las otras subunidades intrínsecas de membrana de la enzima. Hasta el momento, uno de los complejos enzimáticos más estudiados, gracias al empleo de agentes entrecruzadores y a las técnicas de ingeniería genética, es el de E. coli (EF1EFO-ATP sintasa). Los genes que codifican para las subunidades del dominio F1 y FO se encuentran en un único operón, el operón atp o unc (Godinot & Di Pietro, 1986). El complejo enzimático está compuesto por ocho subunidades: cinco en la región F1 con una estequiometría de α3β3γδ y una masa molecular de ∼382 kDa, y tres subunidades en FO con la estequiometría de ab2c10-12 con una masa molecular de ∼148 kDa (Tabla I). Los ocho genes estructurales de este operón están precedidos por un noveno gen, denominado atpI, que se ha encontrado en todos los operones que codifican a las ATPasas bacterianas secuenciados hasta el momento y codifica para una proteína básica hidrofóbica, identificada como Vma21p, que quizá participe en el correcto ensamblaje de la enzima (Deckers-Hebestreit & Altendorf, 1996).
Topología y función de las subunidades del complejo FOSubunidad cLa subunidad c, también conocida como proteolípido, se ha secuenciado a partir de diversas fuentes y posee una serie de características comunes: tiene una masa molecular de 8.3 kDa, y debido a su carácter hidrofóbico es soluble en disolventes orgánicos. Forma un oligómero dispuesto como un anillo embebido en la membrana, en donde cada una de las subunidades que lo componen se pliega como una horquilla con dos alfa hélices unidas por una región polar tipo bucle, expuesta hacia el lado citoplásmico de las bacterias o de la matriz mitocondrial y encima de esta estructura se ubican las subunidades γ/¿ (Deckers-Hebestreit & Altendor, 1996).
Existen varias líneas de evidencia que sugieren la interacción de la subunidad c con estas últimas subunidades. Una de ellas se basa en las mutaciones realizadas en la región tipo bucle, en donde se observó que el acoplamiento entre la subunidad c y la región F1 se ve afectado principalmente en las posiciones Arg41, Asn42 y Pro43 de la subunidad c. Por otra parte, estudios de entrecruzamiento con sustituciones dobles de cisteína en la posición 31 de la subunidad ¿ y en los residuos Ala40, Asn42 y Pro43 de la subunidad c han revelado una posible interacción de estas subunidades. Además, se demostró que en una región que va de la Thr26 a la Gly33 los residuos de cisteína presentes en la subunidad ¿ interactúan con los residuos de cisteína introducidos en las posiciones Ala40, Gln42 y Asp44 de la subunidad c. De igual forma, se determinó una posible interacción de la subunidad γ mediada por su residuo Tyr205 y los aminoácidos Gln42, Pro43 y Asp44 de la región tipo bucle de la subunidad c. Estos resultados sugieren que probablemente la subunidad γ y ¿ interactúan con un conjunto diferente de subunidades del anillo de c, lo que indica que los residuos de esta región se encuentran lo suficientemente expuestos al dominio F1, como se propone en los estudios con agentes entrecruzadores, así como en los análisis de resonancia magnética nuclear (RMN) (Figuras 5 y 6) (Fillingame & Dmitriev, 2002; Altendorf et al., 2000).
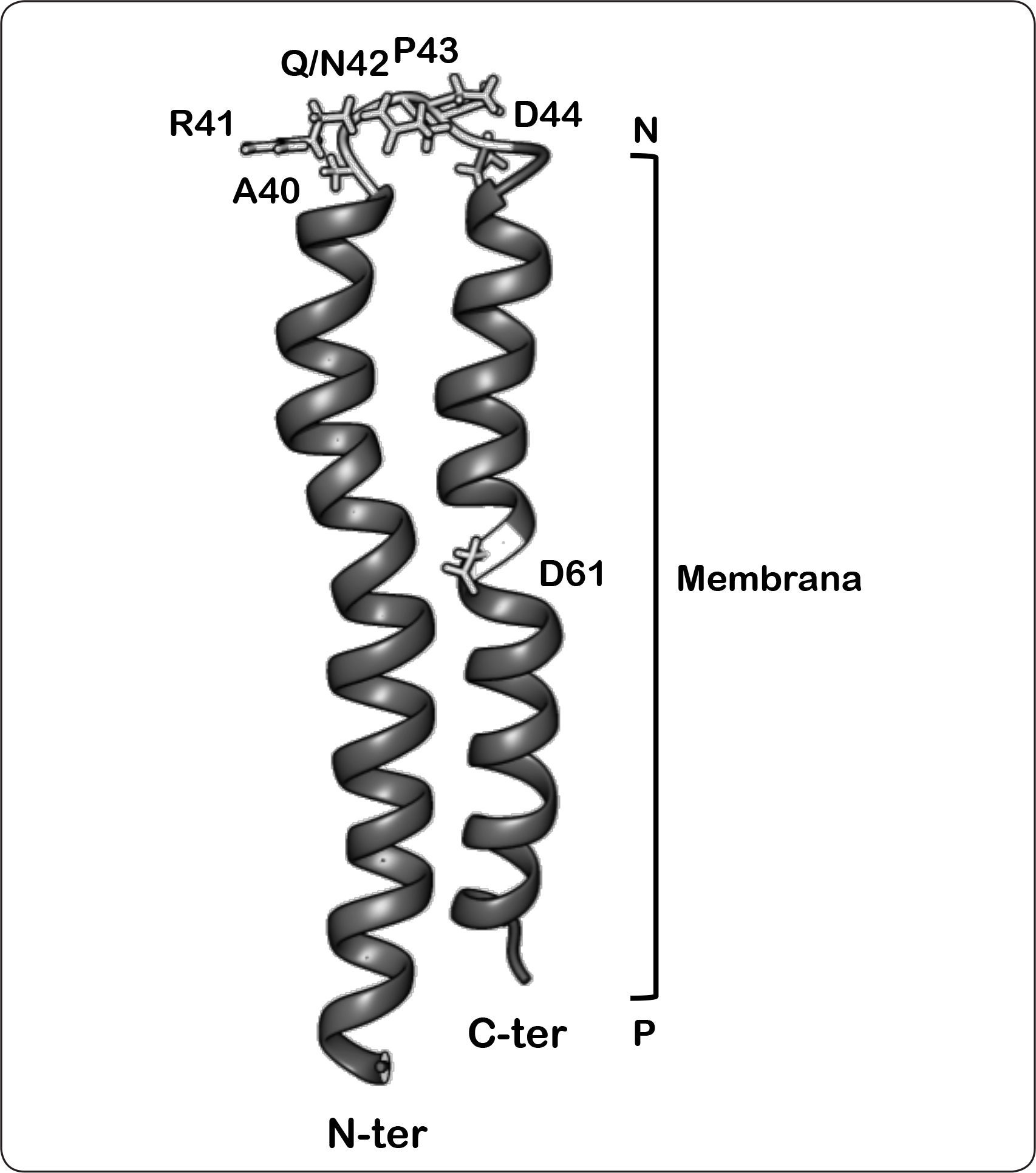
 y lado negativo (N), así como del extremo amino (N-ter, proyectado al frente) y carboxilo terminal (C-ter, proyectado hacia el fondo). Asimismo, se muestran las posiciones de los residuos clave discutidos en el texto. Las coordenadas PDB de la estructura son 1C0V. Modificada de Girvin et al., 1998; Fillingame & Dmitriev, 2002.")
Representación de la estructura tipo horquilla de la subunidad c bacteriana sugerido por estudios de RMN a pH 5. Se indica la orientación de la estructura en la membrana, lado positivo (P) y lado negativo (N), así como del extremo amino (N-ter, proyectado al frente) y carboxilo terminal (C-ter, proyectado hacia el fondo). Asimismo, se muestran las posiciones de los residuos clave discutidos en el texto. Las coordenadas PDB de la estructura son 1C0V. Modificada de Girvin et al., 1998; Fillingame & Dmitriev, 2002.
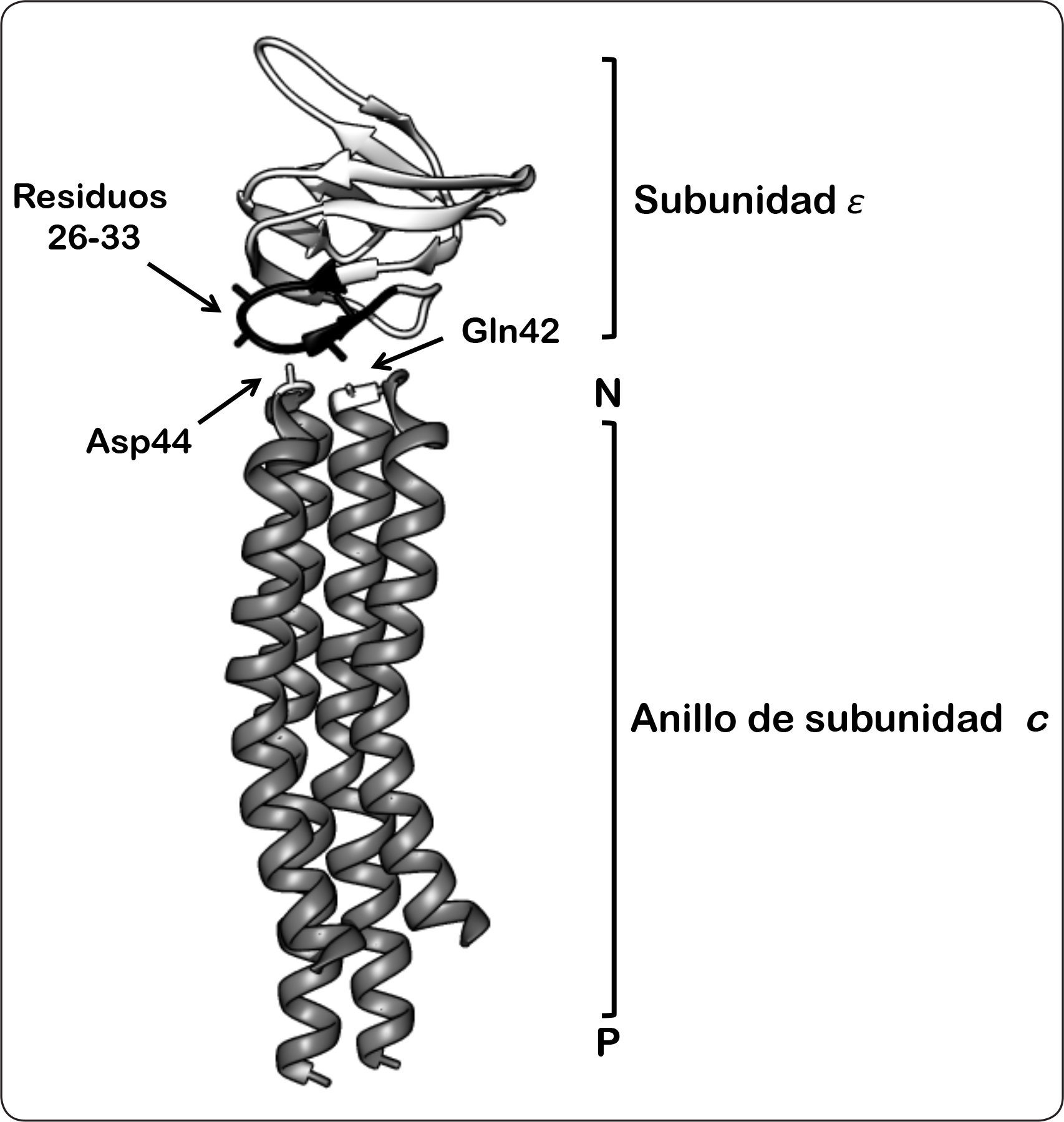
. La región tipo bucle de la subunidad ¿ (residuos 26-33 en color negro), sugiere una posible interacción, ya sea con Gln42 o Asp44 de la subunidad c. P = lado positivo, N = lado negativo. Las coordenadas PDB de la estructura son 5T4O. Modificado de Fillingame & Dmitriev, 2002.")
Modelo de la interacción entre el dominio tipo hoja beta de la subunidad ¿ y la región tipo bucle del anillo de subunidades de c. Esta representación se basa en los estudios de entrecruzamiento de ambas subunidades, discutidas en el texto, así como en los estudios de RMN. El aminoácido Gln42 de la subunidad c se encuentra proyectado al fondo, mientras que hacia el frente se muestra el residuo Asp44 (ambos residuos resaltados en color blanco). La región tipo bucle de la subunidad ¿ (residuos 26-33 en color negro), sugiere una posible interacción, ya sea con Gln42 o Asp44 de la subunidad c. P = lado positivo, N = lado negativo. Las coordenadas PDB de la estructura son 5T4O. Modificado de Fillingame & Dmitriev, 2002.
En la región carboxilo terminal se encuentra el aminoácido Asp61, el cual desempeña un papel preponderante al protonarse aprovechando los iones que fluyen a través del hemicanal de la subunidad a. Esta idea es apoyada por la observación de que la translocación de protones se inhibe al modificar químicamente este residuo con diciclohexilcarbodiimida (DCCD) o al mutarlo por glicina o asparagina. Asimismo, se ha observado que los aminoácidos de la región tipo bucle, Ala40, Arg41, Gln/Asn42 y Pro43, se encuentran altamente conservados en las bacterias, las mitocondrias y los cloroplastos (Figura 5) (Deckers-Hebestreit & Altendorf, 1992).
A partir de imágenes cristalográficas del anillo de subunidades de c de la ATP sintasa de la levadura, así como de imágenes de microscopía de fuerza atómica y de microscopía electrónica de la enzima cloroplastídica y bacteriana, se ha visto que el oligómero de c está compuesto por 10, 14 y 11 subunidades respectivamente. Además, se ha observado que en E. coli dependiendo de las condiciones de crecimiento, la subunidad c puede presentar una estequiometría de 10-12 copias (Yoshida et al., 2001).
De modo que el número de subunidades que conforman el anillo de c puede ser de entre ocho a quince copias de la subunidad c, y en consecuencia este oligómero puede variar de acuerdo a la especie. Por ejemplo, se ha observado que las ATP sintasas de los eucariotas tienen anillos pequeños, mientras que las enzimas de los procariotas y los cloroplastos generalmente presentan anillos grandes, como en las cianobacterias que presentan un oligómero de 15 subunidades (Figura 7) (Nesci et al., 2016).
![Comparación de la estructura del anillo de subunidades de c de diferentes organismos. Se muestra la vista superior y lateral de cada estructura oligomérica con su disposición espacial y el número de subunidades que componen a cada oligómero. Las imágenes de cristalografía de rayos X corresponden a: Bos taurus (2XND) [c8], Saccharomyces cerevisiae (2XOK) [c10], Acetobacter woodii (no homomérica, la subunidad c doble se resalta en negro, 4BEM) [c’9+c”1], Ilyobacter tartaricus (1YCE) [c11], Bacillus pseudofirmus (2X2V) [c13], y la cloroplastídica de Spinacia oleracea (2W5J) [c14]. Los anillos de subunidad c de I. tartaricus y A. woodii unen iones Na+, mientras que los de B. taurus, S. cerevisiae, B. pseudofirmus, y S. oleracea unen iones H+ (número de acceso PDB entre paréntesis). Modificado de Nesci et al., 2016.](https://static.elsevier.es/multimedia/1405888X/0000002000000002/v1_201706150025/S1405888X17300049/v1_201706150025/es/main.assets/gr7.jpeg?xkr=ue/ImdikoIMrsJoerZ+w997EogCnBdOOD93cPFbanNcNGSBFqzjIokZJFmIUKi41CTrJidfd4C+mTEouRVcDMbVgqpp/EkEG/e7vkw0PmM8YrEZFtLFJ7tJLyvsqDi/nsWx8IK0Ef82Kbyqli+quO7fCu1blR8658S3bv8G940gRLkqcbllVzr5BWqOMtk5CxhdUt0WALQLFxfJPypYf26He92rvJkUIofgbKlQD2MXGMoPJX/Kt1TevhZCpC+y1wnRV55EKlNnpSdknzl2Hmw== "Comparación de la estructura del anillo de subunidades de c de diferentes organismos. Se muestra la vista superior y lateral de cada estructura oligomérica con su disposición espacial y el número de subunidades que componen a cada oligómero. Las imágenes de cristalografía de rayos X corresponden a: Bos taurus (2XND) [c8], Saccharomyces cerevisiae (2XOK) [c10], Acetobacter woodii (no homomérica, la subunidad c doble se resalta en negro, 4BEM) [c’9+c”1], Ilyobacter tartaricus (1YCE) [c11], Bacillus pseudofirmus (2X2V) [c13], y la cloroplastídica de Spinacia oleracea (2W5J) [c14]. Los anillos de subunidad c de I. tartaricus y A. woodii unen iones Na+, mientras que los de B. taurus, S. cerevisiae, B. pseudofirmus, y S. oleracea unen iones H+ (número de acceso PDB entre paréntesis). Modificado de Nesci et al., 2016.")
Comparación de la estructura del anillo de subunidades de c de diferentes organismos. Se muestra la vista superior y lateral de cada estructura oligomérica con su disposición espacial y el número de subunidades que componen a cada oligómero. Las imágenes de cristalografía de rayos X corresponden a: Bos taurus (2XND) [c8], Saccharomyces cerevisiae (2XOK) [c10], Acetobacter woodii (no homomérica, la subunidad c doble se resalta en negro, 4BEM) [c’9+c”1], Ilyobacter tartaricus (1YCE) [c11], Bacillus pseudofirmus (2X2V) [c13], y la cloroplastídica de Spinacia oleracea (2W5J) [c14]. Los anillos de subunidad c de I. tartaricus y A. woodii unen iones Na+, mientras que los de B. taurus, S. cerevisiae, B. pseudofirmus, y S. oleracea unen iones H+ (número de acceso PDB entre paréntesis). Modificado de Nesci et al., 2016.
Las estructuras cristalográficas del anillo de subunidades de c sugieren que la estequiometría del anillo es una adaptación para funcionar de manera óptima bajo las condiciones bioenergéticas predominantes. En consecuencia, cuando el potencial eléctrico de la membrana (Δϕ, gradiente eléctrico) es la fuerza dominante de conducción, por ejemplo en las mitocondrias, el tamaño del anillo es pequeño. Por el contrario, en los cloroplastos, en donde una gran diferencia en la concentración de protones (ΔpH, gradiente químico) se acompaña de una baja magnitud del valor Δϕ la ATPasa presenta un anillo formado por un gran número de subunidades c, como en las cianobacterias con estequiometrías de hasta 15 copias. Al parecer esta diferencia en el número de subunidades c compensa la baja magnitud del valor Δϕ (Nesci et al., 2016).
Dado que una rotación completa del rotor de la ATP sintasa (360°) produce tres moléculas de ATP y que en cada rotación del anillo de c se translocan igual número de iones (protones o sodio) que subunidades que lo componen, el costo bioenergético para la ATP sintasa equivale al número de subunidades c dividido entre tres. Entonces, para la ATP sintasa mitocondrial de bovino, con un anillo de c de ocho subunidades es 8/3 ó 2.7 protones por molécula de ATP. Siguiendo esta misma lógica, en otros organismos como las bacterias o los cloroplastos de plantas verdes, en donde se han observado tamaños de anillo de 10, 11, 13, 14 y 15 subunidades c (Figura 7), el costo bioenergético para la ATP sintasa sería de 4.3, 4.7, 5.3 y 6 protones por ATP producido respectivamente (incluyendo el H+ extra que se requiere para la salida del ATP) (Walker, 2013).
Subunidad b bacterianaEn E. coli la subunidad b (17.3 kDa) es una proteína anfipática que se ha dividido en dominios funcionales. El primero de ellos es un dominio que atraviesa la membrana, formado por los 33 aminoácidos del extremo amino terminal, cuya función es anclar esta proteína a la bicapa lipídica. Otro dominio está compuesto por los residuos 53-122, el cual se ha mostrado que es el responsable de la interacción de las subunidades b para formar el dímero a través de los aminoácidos hidrofóbicos situados en esta región. Además, este segmento de la subunidad b se ha asociado a la formación de los dímeros de la ATP sintasa. Asimismo, en el extremo carboxilo se encuentra el dominio de unión a la región F1 y la interacción se lleva a cabo por una de las subunidades b del dímero con la subunidad δ, mientras que el otro monómero lo hace con un par de subunidades α/β, apoyando la idea de que la subunidad b es el componente principal del estator del complejo enzimático. En este sentido, se ha observado que la presencia de la subunidad b es importante para el correcto ensamblaje de los dominios F1 y FO. De igual forma, se ha determinado que el dímero de subunidades b es capaz de interactuar con las subunidades del sector FO y de esta forma los segmentos transmembranales de la subunidad b, aunado a la subunidad a y el anillo de subunidad c, son suficientes para que se forme el canal que permite la translocación de protones (Dmitriev et al., 1999; Welch et al., 2008; Aris et al., 1985).
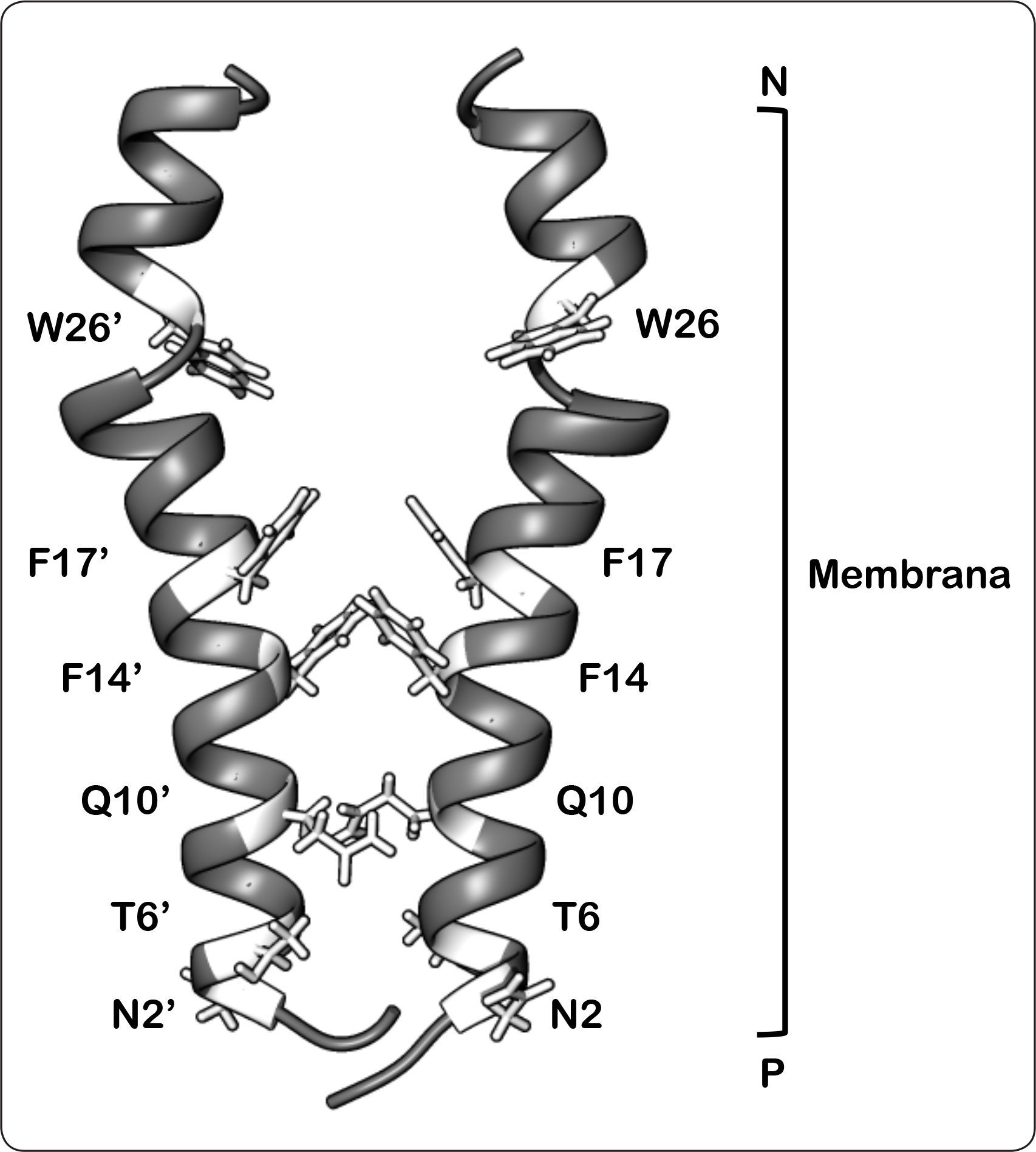
Considerando lo anterior, se ha propuesto un modelo de interacción del dominio membranal de la subunidad b, el cual se basa en los resultados obtenidos mediante el uso de entrecruzadores y estudios de RMN (Dmitriev et al., 1999). Este modelo propone que las interacciones de los segmentos transmembranales se da entre los anillos aromáticos de los aminoácidos Phe14-Phe17’, en donde cada residuo corresponde a un monómero de subunidad b, así como entre Phe17-Phe14’ con una distancia de interacción de 5 y 5.8Å, respectivamente. Además, las hélices transmembranales están dispuestas en un ángulo de 23° y las interacciones de la interfaz de cada monómero principalmente son contactos de tipo Van der Waals, puesto que se compone por las cadenas laterales de los residuos Asn2, Thr6, Gln10, Phe14 y Phe17 (Figura 8) (Dmitriev et al., 1999). Por otro lado, se ha observado que el residuo Arg36 se encuentra altamente conservado en las ATP sintasas bacterianas, lo que sugiere su probable participación en la interacción a nivel membranal (Welch et al., 2008).

Modelo de interacción de los dominios transmembranales de la subunidad b. Las cadenas laterales de los aminoácidos tratados en el texto se muestran en color. Las cadenas laterales de los residuos de Phe14, Phe17, y Phe14’, Phe17 interactúan en un grupo hidrofóbico. De igual forma, los residuos Asn2, Thr6, Gln10 y Asn2’, Thr6’, Gln10’ establecen contactos tipo Van der Waals. Los anillos aromáticos de Trp26 y Trp26’ se proyectan hacia la parte delantera y trasera de la estructura, respectivamente. P = lado positivo, N = lado negativo. Modificado de Dmitriev et al., 1999.
En la levadura, la subunidad 4 está formada por 209 aminoácidos y tiene una masa molecular estimada de 23.25 kDa. Esta proteína es homóloga a la subunidad b de la enzima bacteriana y a la subunidad b de la mitocondria de bovino (Tabla I). De acuerdo con métodos de predicción de la estructura secundaria, esta subunidad está compuesta por dos segmentos altamente hidrofóbicos en la región amino terminal y un sector carboxilo terminal con carácter anfipático. A partir de ensayos de proteólisis controlada con partículas submitocondriales, se concluyó que la subunidad 4, al igual que su homólogo en bacteria, se localiza en la interfaz entre los dominios F1 y FO (Paul et al., 1989).
Por otra parte, se ha propuesto que la subunidad 4 desempeña una función importante durante la biogénesis del complejo V. Se ha visto que la interrupción del gen ATP4 afecta la organización de los supercomplejos mitocondriales, así como la actividad de la citocromo oxidasa, disminuyendo su actividad aproximadamente una quinta parte en comparación con la cepa silvestre de la levadura (Paul et al., 1989). En este sentido, estudios previos con mutantes en las cuales se eliminó el primer segmento transmembranal, muestran un fenotipo mutante nulo, al igual que como sucede con las mutantes de los genes TIM11 o ATP20, es decir, un aumento en el tiempo de duplicación, carencia de formas diméricas de la ATP sintasa y una morfología mitocondrial alterada, aunado a la carencia de la subunidad g. Por el contrario, cuando se realizaron ensayos con mutantes que cuentan con los primeros 18 aminoácidos del N terminal lo anterior se revirtió. Estos resultados sugieren que al eliminar el primer cruce transmembranal de la subunidad 4 se pierde la interacción con la subunidad g, dando lugar al efecto observado (Soubannier et al., 2002).
A partir de la información obtenida con las mutantes y los entrecruzadores, se ha sugerido un modelo topológico de la subunidad 4 (Figura 9), en donde se muestran los contactos entre la alfa hélice membranal de la subunidad 6, los dos segmentos hidrofóbicos de la subunidad 4 y el único cruce transmembranal de las subunidades f, i y g (estas dos últimas ubicadas detrás de la subunidad f). En la parte superior, se pueden observar las interacciones de la subunidad 4 con OSCP y la subunidad beta (Velours et al., 2000).

Modelo de topología de la subunidad 4 de levadura. Los cilindros representan alfa hélices. De estos, los grises representan a la subunidad 4. Los círculos negros son los residuos de cisteína que fueron introducidos por mutagénesis sitio dirigida. El residuo 209 es el último de la subunidad 4. P = lado positivo, N = lado negativo. Modificado de Velours et al., 2000.
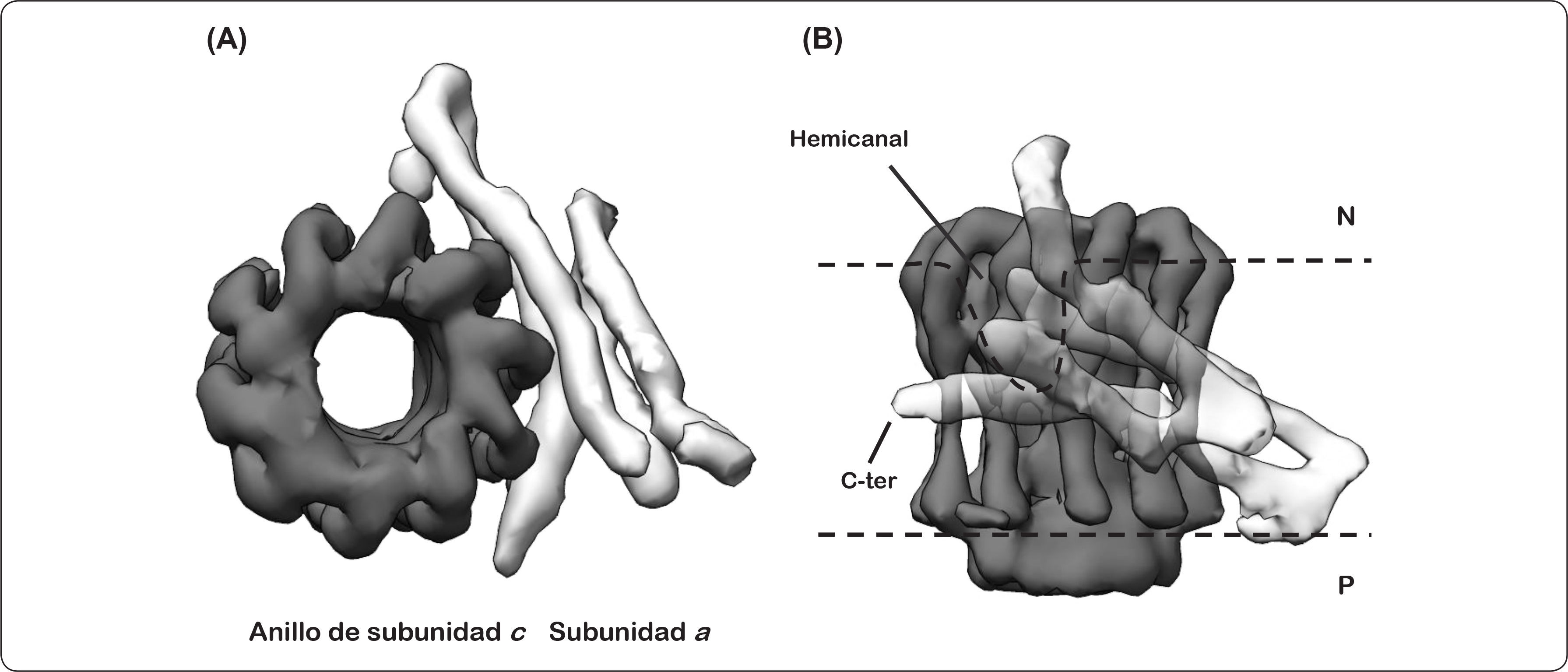
La subunidad a es una proteína extremadamente hidrofóbica con una masa molecular de 30.132 kDa, la cual se ha explorado por más de un centenar de sustituciones de monocisteína. Estudios previos de esta subunidad han mostrado que presenta cinco segmentos transmembranales, con el extremo amino y carboxilo terminal orientados hacia el lado citoplásmico bacteriano (lado negativo). De igual forma, se determinaron los aminoácidos clave en la translocación de protones y que están altamente conservados, como la Arg210 que se encuentra cercana al centro de la bicapa lipídica, algunos otros aminoácidos como la His245 y el Glu219, presentes en el lado periplásmico (lado positivo), y el Glu196 expuesto hacia el citoplasma. Por otra parte, se observó que los últimos diez residuos del carboxilo terminal son prescindibles para la función de la proteína. La subunidad a tiene dos bucles citoplásmicos, el primero de ellos contiene alrededor de 40 aminoácidos (60-103), mientras que el segundo presenta aproximadamente 35 aminoácidos (160-206). Adicionalmente, se ha propuesto que probablemente el primero de estos bucles interacciona con una o dos de los subunidades b, asimismo los segmentos transmembranales cuarto y quinto de la subunidad a están en contacto con dos subunidades c (Figura 10, panel A) (Vik & Ishmukhametov, 2005). Estudios recientes sugieren que la subunidad a presenta cuatro segmentos helicoidales horizontales y un cruce transmembranal vertical, formando dos hemicanales a través de los cuales se translocan los iones (Figura 10, panel B) (Nesci et al., 2015; Allegretti et al., 2015; Zhou et al., 2015).
 Imágenes obtenidas por crío-microscopía electrónica (7.0Å de resolución, no. de acceso EMD 2852) de un corte transversal a nivel de membrana del dominio FO de la ATP sintasa de Polytomella sp., en donde se muestra a la izquierda el anillo de subunidad c (negro) y a la derecha la densidad electrónica de las hélices horizontales membranales de la subunidad a (gris). En este modelo no se representa al amino terminal. (B) Vista lateral de (A), en donde se muestra al anillo de subunidad c al fondo (negro), y hacia el frente los segmentos alfa-helicoidales atravesando la membrana de manera horizontal (cuatro segmentos en gris). Se señala tanto el hemicanal formado por la interacción de ambas subunidades, así como la región carboxilo (C-ter) de la subunidad a. P = lado positivo, N = lado negativo. Modificado de Allegretti et al., 2015; Kühlbrandt & Davies, 2016.")
Modelo estructural del anillo de subunidades c-subunidad a. (A) Imágenes obtenidas por crío-microscopía electrónica (7.0Å de resolución, no. de acceso EMD 2852) de un corte transversal a nivel de membrana del dominio FO de la ATP sintasa de Polytomella sp., en donde se muestra a la izquierda el anillo de subunidad c (negro) y a la derecha la densidad electrónica de las hélices horizontales membranales de la subunidad a (gris). En este modelo no se representa al amino terminal. (B) Vista lateral de (A), en donde se muestra al anillo de subunidad c al fondo (negro), y hacia el frente los segmentos alfa-helicoidales atravesando la membrana de manera horizontal (cuatro segmentos en gris). Se señala tanto el hemicanal formado por la interacción de ambas subunidades, así como la región carboxilo (C-ter) de la subunidad a. P = lado positivo, N = lado negativo. Modificado de Allegretti et al., 2015; Kühlbrandt & Davies, 2016.
Este último hallazgo, la disposición casi horizontal de los segmentos alfa helicoidales hidrofóbicos de la subunidad a con respecto a la membrana, fue una sorpresa, ya que la evidencia experimental obtenida en las últimas dos décadas sugería que estas hélices se encontrarían dispuestas de manera perpendicular a la membrana, similar a las alfa hélices que componen el anillo de subunidades de c. Como se puede apreciar en el mapa de densidad electrónica para la F-ATPasa de Polytomella sp. (Figura 10), el hemicanal está formado por una interfaz entre la subunidad a y el oligómero de subunidades de c compuesta en su mayor parte por los residuos polares conservados, dando lugar a un canal acuoso a través del cual se translocan los protones (Kühlbrandt & Davies, 2016).
Mecanismo de la translocación de los protonesLas dos funciones opuestas de la F1FO-ATP sintasa, síntesis (ATP sintasa) e hidrólisis de ATP (ATPasa), se basan en la capacidad del rotor, embebido en la membrana, de girar en dos direcciones contrarias. En condiciones fisiológicas, el mecanismo enzimático del complejo es la síntesis de ATP, este proceso se da a través de un acoplamiento quimio-mecánico que implica un movimiento de rotación del motor (FO) impulsado por la fuerza protón motriz (Δp), dando lugar a la translocación de protones del lado positivo (P) de la membrana hacia el lado negativo (N) de la misma. Por el contrario, bajo condiciones anaerobias, bajo Δp, la enzima hidroliza el ATP para mover los protones en el sentido opuesto, sentido anti horario visto desde el dominio F1, restableciendo así el gradiente electroquímico de protones. El equilibrio termodinámico entre el potencial de fosforilación (ΔGp) del ADP y el Δp determina cuál de estas dos actividades enzimáticas, síntesis o hidrólisis de ATP, se lleva a cabo por la ATP sintasa/ATPasa (Nesci et al., 2015).
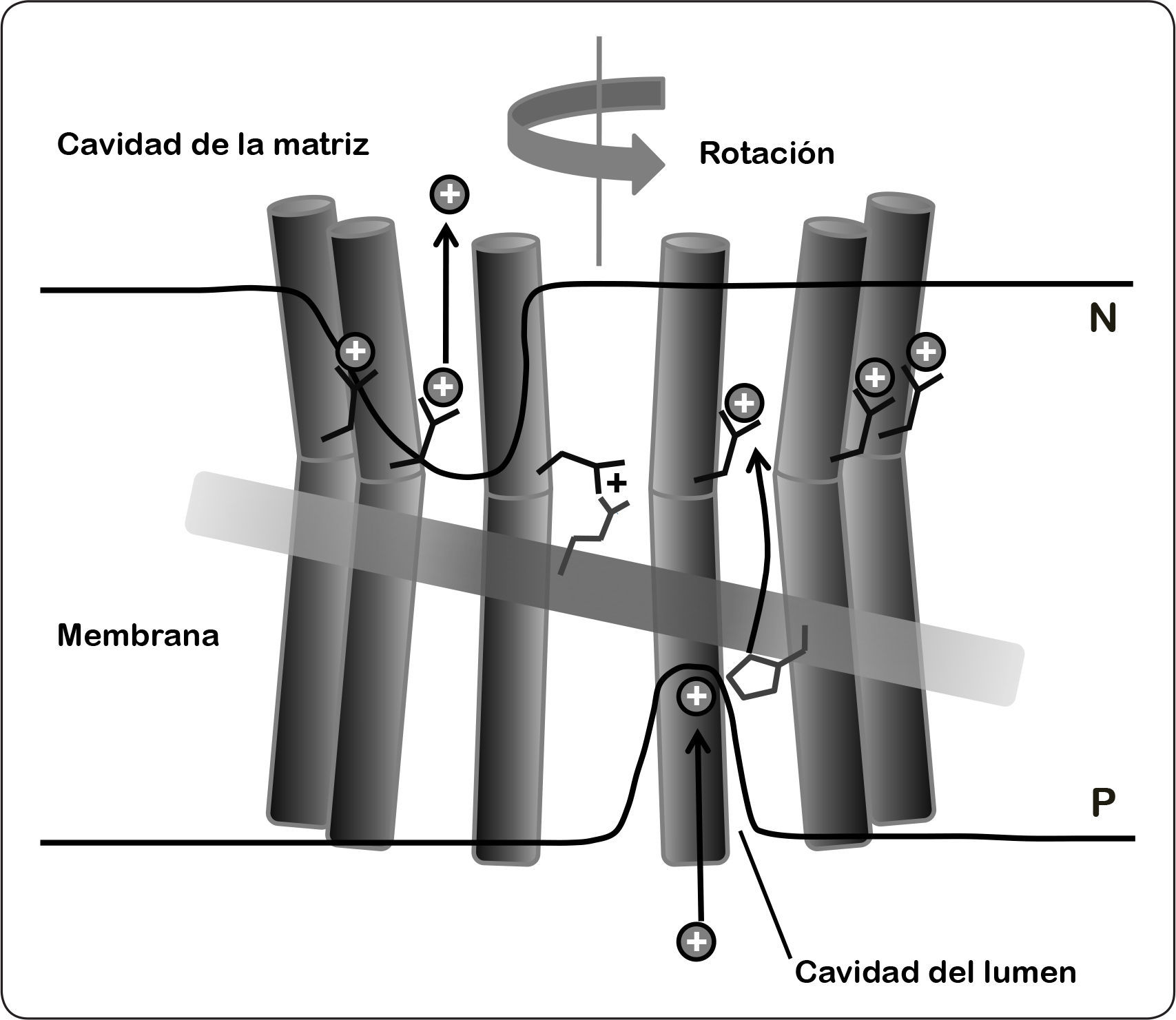
Los constituyentes fundamentales en la generación de la fuerza de torque son la subunidad a y el anillo de subunidades c. Estas subunidades cuentan con elementos que permiten la translocación de iones: la carga positiva de un grupo guanidinio de la Arg (altamente conservado) en la subunidad a y los grupos carboxilo de los residuos de Asp o Glu (dependiendo de la especie) del anillo de c, que permiten la unión de los protones gracias a su carga negativa. Estos sitios de protonación se encuentran cercanos a los hemicanales y son accesibles tanto del lado N como del P de la bicapa lipídica. En la dinámica de rotación del motor FO, cuando el Δp es lo suficientemente alto, el hemicanal que se encuentra expuesto en el lado P de la membrana constituye la vía de entrada de los protones a los grupos carboxilo del oligómero de c. Una vez protonados, la subunidad c se vuelve menos polar y por lo tanto es capaz de entrar a la bicapa lipídica, manteniendo el residuo orientado hacia el centro del anillo del rotor, es decir, pasa de una conformación abierta a una cerrada, lo cual es favorecido energéticamente. Al mismo tiempo, una subunidad c con un grupo carboxilo neutralizado está expuesta en la cara del hemicanal que mira hacia el lado negativo de la membrana. Este entorno más hidrofílico favorece que el grupo carboxilo cambie a una conformación abierta que apunta hacia la subunidad a. En estas condiciones, el pH básico en el lado N de la membrana y la carga positiva de la Arg, dan lugar a reordenamientos estructurales que disminuyen el pKa de este grupo permitiendo el desprendimiento de protones. En consecuencia, el carboxilato desprotonado se estabiliza en la conformación abierta a través de puentes salinos con el aminoácido Arg, cuyo pKa mantiene al grupo guanidinio protonado con carga positiva. De esta forma, cuando un nuevo protón se enfrenta al hemicanal en el lado P, el sitio de unión carboxilato nuevamente cambia a la forma protonada, acompañado de un cambio en la conformación para iniciar un nuevo ciclo de rotación (Figura 11) (Nesci et al., 2015).
 de la hélice horizontal membranal de la subunidad a (rectángulo horizontal) expuesta al lado de la cavidad luminal. Las flechas indican el canal propuesto mediante el cual los protones se abren paso a través de la bicapa lipídica. Los iones H+ se unen a la conformación abierta del glutamato cargado (negro) de la subunidad c (cilindros en color oscuro). El mecanismo de protonación (cavidad del lúmen), y desprotonación (cavidad de la matriz) dan lugar a la rotación del anillo de subunidad c (la flecha superior gris indica el sentido del giro). Este movimiento en sentido opuesto se evita por la presencia de arginina (residuo ubicado en la parte superior izquierda del rectángulo), ésta se encuentra a 2.5 vueltas de hélice (13.5Å) por debajo del aminoácido glutámico. Esta distancia coincide con el espacio entre los sitios de protonación adyacentes en el anillo de subunidades de c de cualquier estequiometría conocida. De este modo, los residuos de arginina e histidina pueden interaccionar al mismo tiempo con dos hélices adyacentes de subunidad c como se requiere para la translocación de protones. P = lado positivo, N = lado negativo. Modificado de Kühlbrandt & Davies, 2016.")
Modelo del proceso de translocación de los protones. Los protones, representados como un círculo negro con carga positiva, acceden al hemicanal al interaccionar con el residuo de histidina (residuo ubicado en la parte inferior derecha del rectángulo) de la hélice horizontal membranal de la subunidad a (rectángulo horizontal) expuesta al lado de la cavidad luminal. Las flechas indican el canal propuesto mediante el cual los protones se abren paso a través de la bicapa lipídica. Los iones H+ se unen a la conformación abierta del glutamato cargado (negro) de la subunidad c (cilindros en color oscuro). El mecanismo de protonación (cavidad del lúmen), y desprotonación (cavidad de la matriz) dan lugar a la rotación del anillo de subunidad c (la flecha superior gris indica el sentido del giro). Este movimiento en sentido opuesto se evita por la presencia de arginina (residuo ubicado en la parte superior izquierda del rectángulo), ésta se encuentra a 2.5 vueltas de hélice (13.5Å) por debajo del aminoácido glutámico. Esta distancia coincide con el espacio entre los sitios de protonación adyacentes en el anillo de subunidades de c de cualquier estequiometría conocida. De este modo, los residuos de arginina e histidina pueden interaccionar al mismo tiempo con dos hélices adyacentes de subunidad c como se requiere para la translocación de protones. P = lado positivo, N = lado negativo. Modificado de Kühlbrandt & Davies, 2016.
Con respecto al mecanismo de translocación de iones, se ha observado un retraso en el movimiento de rotación de 50-175μs en la F-ATPasa de E. coli. Se ha propuesto que esta pausa es resultado de la interposición de las subunidades c adyacentes. Sin embargo, estudios recientes sugieren que este movimiento se debe a la formación de un puente salino, entre los residuos Glu196 de la subunidad a y Arg50 de la subunidad c. Asimismo, esta pausa podría ser determinante en la dirección del giro del anillo de subunidades c en el sentido de la síntesis o de la hidrólisis de ATP (Martin et al., 2014; Martin et al., 2015).
Otras subunidades del domino membranal de la F1FO-ATP sintasa mitocondrialSin considerar su origen, la F1FO-ATP sintasa mitocondrial también se compone de dos grandes dominios funcionales, el sector hidrofílico F1 y un dominio membranal FO, unidos por un tallo rotor central y un brazo periférico. Las ATPasas mitocondriales del bovino y la levadura son las más estudiadas y las mejor caracterizadas en términos de estructura y función. La mayoría de las subunidades del complejo enzimático de la levadura son proteínas homólogas a la enzima de bovino, mientras que algunas otras son especie específicas (Lee et al., 2015).
Con respecto a lo anterior, la ATP sintasa monómerica de levadura presenta 14 subunidades: α(3), β(3), γ(1), δ(1), ¿(1), la subunidad 6 ó a(1), b(1), subunidad 9 o c(10), d(1), f(1), h(1), i ó k(1), subunidad 8(1), y OSCP(1) con la estequiometría asignada (Tabla I). El peso molecular estimado de este complejo enzimático es de entre 572.759 kDa a 573.067 kDa considerando la formilación del amino terminal de las subunidades 8 y 9. En la ATPasa dimérica se encuentran tres proteínas adicionales, subunidades i(k), e y g, conocidas también como subunidades específicas de dimerización, puesto que sólo se han observado en el dímero de la ATP sintasa. El peso molecular aproximado de la forma dimérica es de 1,207.916 kDa, asumiendo una estequiometría 1:1 para las subunidades de dimerización, o de 1,208.616 kDa tomando en cuenta nuevamente, la formilación de las subunidades 8 y 9, así como la acetilación de la subunidad e (Tabla I) (Wittig & Schägger, 2008).
Por el contrario, en la enzima de bovino las subunidades e y g se encuentran asociadas fuertemente a la ATPasa y se han podido aislar del monómero de la enzima. La forma monómerica está compuesta por 15 proteínas, subunidad α(3), β(3), γ(1), δ(1), ¿(1), subunidad 6 ó a(1), b(1), subunidad 9 ó c(10), d(1), e(1), f(1), g(1), F6(1), subunidad 8 ó A6L(1), y OSCP(1). Considerando la estequiometría asignada, la masa molecular de este complejo es de 583.422 kDa ó 583. 573 kDa incluyendo las modificaciones postraduccionales del amino terminal. Recientemente, se han encontrado dos proteínas asociadas a la F-ATPasa mitocondrial de bovino y rata, la subunidad MLQ y la proteína AGP (Tabla I) (Wittig & Schägger, 2008).
Topología y función de las subunidades del complejo FOSubunidad 8También llamada proteína A6L en bovino, es considerada un proteolípido debido a su solubilidad en disolventes orgánicos (cloroformo/metanol). Se ha dividido en tres dominios estructurales y funcionales: un dominio amino terminal conservado, una región hidrofóbica central y un extremo carboxilo terminal que presenta tres aminoácidos con carga positiva altamente conservados. Se ha descrito que los residuos 15-35 del dominio amino terminal se localizan en la membrana, orientados hacia el espacio intermembranal y debido a la conservación de esta región, se ha propuesto que estos aminoácidos son importantes para la correcta importación de la proteína. Por otro lado, estudios previos sugieren que los tres residuos con carga positiva del dominio carboxilo terminal son necesarios para el correcto ensamblaje de esta subunidad en el sector FO. La eliminación de los aminoácidos Arg37, Arg42 y Lys47 tiene un impacto severo tanto en el ensamblaje de esta proteína, como en su importación in vitro y actividad de ATPasa in vivo (Devenish et al., 1992; Grasso et al., 1991).
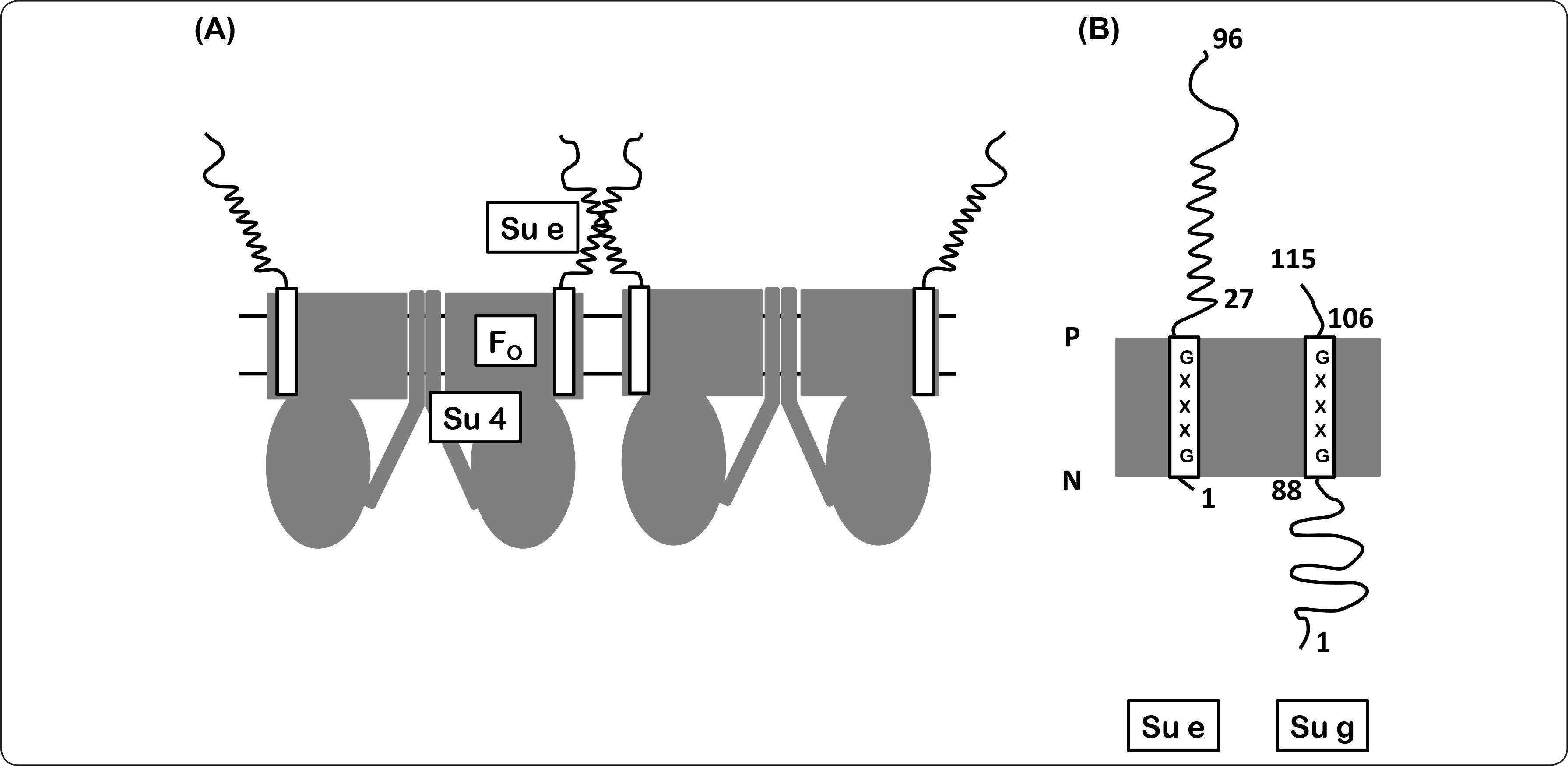
Subunidad eEsta subunidad es una proteína integral anclada a la membrana por un segmento hidrofóbico en el extremo amino. La subunidad e se compone de 96 aminoácidos (10.744 kDa), esta proteína presenta una topología N-adentro (matriz mitocondrial) C-fuera (espacio intermembranal) y tiene una estequiometría de 2 subunidades por cada subunidad γ de la ATPasa. La región amino terminal presenta el motivo GxxxG (Gly-15-Gly19), el cual se ha descrito participa en la formación de homo- o hetero-dímeros membranales (Everard-Gigot et al., 2005; Brunner et al., 2002). En este sentido, en S. cerevisiae se ha propuesto que la proteína Mgm1p se requiere para la regulación y estabilidad de la misma (Amutha et al., 2004). La subunidad e y g de levadura están involucradas en los procesos de dimerización/oligomerización de la enzima (Figura 12). La eliminación de estas proteínas por ingeniería genética trae como consecuencia una morfología mitocondrial con pérdida de crestas, numerosas digitaciones y estructuras de tipo cebolla, lo que sugiere un vínculo entre la oligomerización del complejo enzimático y la arquitectura de las crestas mitocondriales (Arselin et al., 2003; Arselin et al., 2004).
 En este modelo, se representa la interacción del dímero de la subunidad e y la interacción de las subunidades e+g (hélices con un segmento membranal) estabilizando los contactos del sector FO, además de la interacción de la subunidad 4 en cada uno de los monómeros de la enzima. Por simplicidad, en el modelo sólo se representa una hélice que corresponde al homodímero e+e y al heterodímero e+g. (B) Topología de membrana de las subunidades e y g. Se indica la orientación de cada subunidad de acuerdo al número de aminoácido correspondiente, igualmente se resalta el dominio GxxxG. P = lado positivo, N = lado negativo. Modificado de Brunner et al., 2002.")
Modelo de la participación de las subunidades 4, e y g en el dímero de la ATP sintasa. (A) En este modelo, se representa la interacción del dímero de la subunidad e y la interacción de las subunidades e+g (hélices con un segmento membranal) estabilizando los contactos del sector FO, además de la interacción de la subunidad 4 en cada uno de los monómeros de la enzima. Por simplicidad, en el modelo sólo se representa una hélice que corresponde al homodímero e+e y al heterodímero e+g. (B) Topología de membrana de las subunidades e y g. Se indica la orientación de cada subunidad de acuerdo al número de aminoácido correspondiente, igualmente se resalta el dominio GxxxG. P = lado positivo, N = lado negativo. Modificado de Brunner et al., 2002.
Esta proteína posee 115 aminoácidos y su extremo amino se encuentra orientado hacia la matriz mitocondrial y su corto dominio carboxilo está ubicado en el espacio intermembranal. Se ha identificado un motivo GxxxG (Gly101-Gly105) altamente conservado en distintos organismos, que se encuentra en la región que atraviesa la membrana y se ha demostrado que éste es el responsable de la participación de la subunidad g en la dimerización de la enzima (Bustos & Velours, 2005; Saddar & Stuart, 2005). Estudios anteriores empleando agentes entrecruzadores, mostraron que la formación del heterodímero e+g es específico del dímero de ATPasa, mientras que los homodímeros e+e y g+g se encuentran únicamente en las formas oligoméricas de la enzima (Fronzes et al., 2006). Considerando lo anterior, se ha propuesto un modelo de interfaz, en donde dos subunidades 4 del complejo enzimático, una subunidad por monómero, participan en la formación de dos interfaz: una implica el dímero de la subunidad 4 y la otra corresponde a las subunidades e y g. Estas superficies de contacto no son independientes, puesto que la eliminación del extremo amino terminal de la subunidad 4 conduce a la pérdida de la subunidad g y de las formas oligoméricas de la enzima, mientras que el mismo efecto se presenta tras la pérdida de la subunidad e ó g (Figura 12) (Bustos & Velours, 2005).
Subunidad fLa subunidad f es una proteína de 101 aminoácidos, que está codificada en el gen ATP17 con un marco abierto de lectura de 303 pb. La subunidad madura se compone de 95 aminoácidos con una masa de 10.567 kDa. Es una proteína básica con un pI estimado de 9.98. Presenta 19 residuos básicos y 11 residuos ácidos. El análisis del perfil de hidrofobicidad muestra un segmento membranal putativo de 18 aminoácidos cercano al extremo carboxilo terminal (Spannagel et al., 1997). Empleando agentes entrecruzadores, se determinó que presenta una topología N-dentro C-fuera. Estudios previos realizados con mutantes del gen ATP17, mostraron que al eliminar los últimos 28 aminoácidos de la secuencia, incluyendo el segmento membranal, la actividad de la ATPasa disminuía dos veces en comparación a la cepa silvestre (Roudeau et al., 1999).
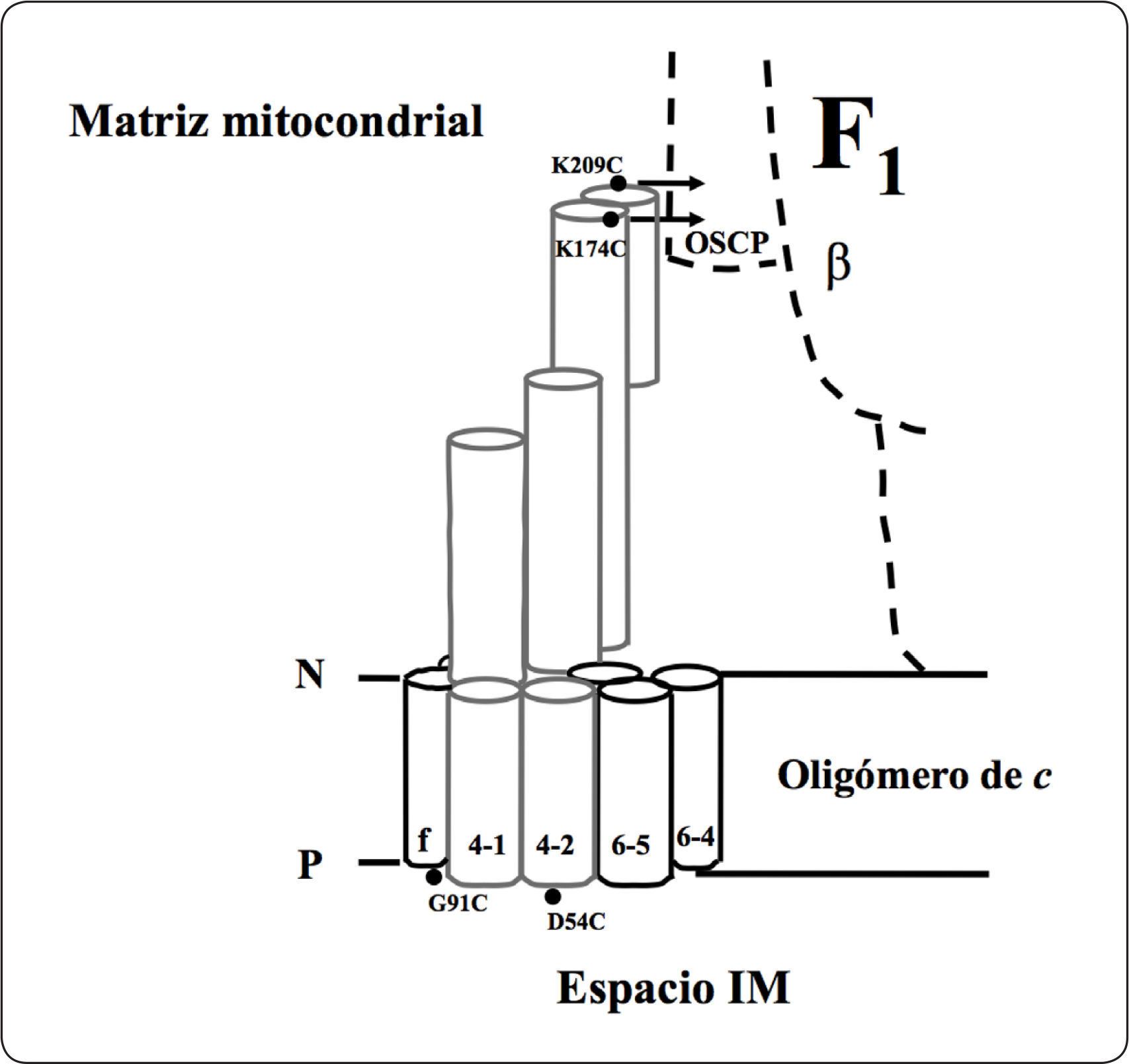
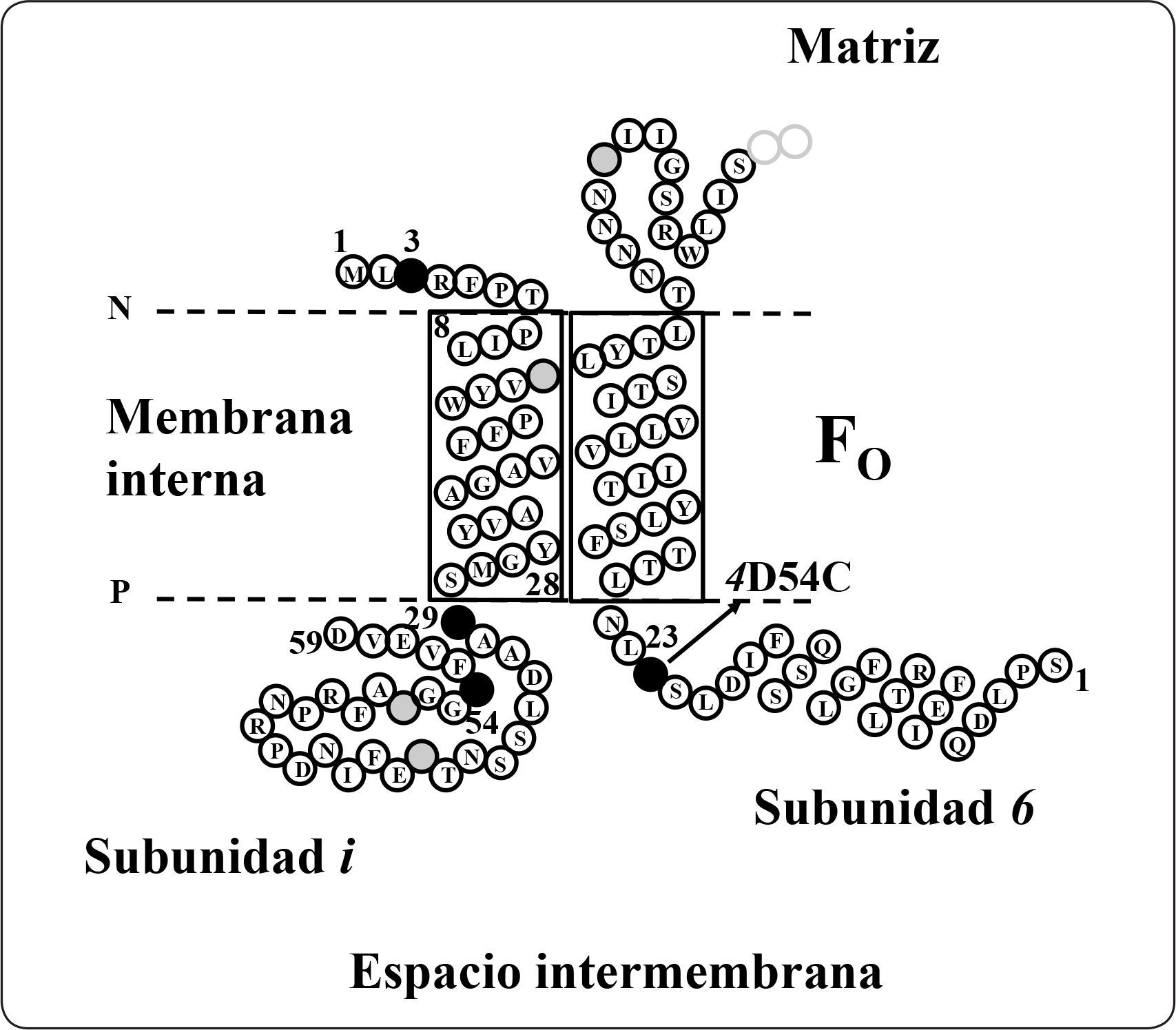
Subunidades i/j, k, lLa subunidad i, también llamada j, está codificada en el gen ATP18, tiene 59 aminoácidos y una masa molecular estimada de 6.687 kDa. Es una proteína de la membrana interna mitocondrial que presenta una topología N-dentro C-fuera. Análisis de alineamientos múltiples de secuencia determinaron que tiene homólogos en Schizosaccharomyces pombe (EMBL número de acceso Z99753), Neurospora crassa (GenBank número de acceso AI329387) y Caenorhabditis elegans (GenBank con números de acceso AF067943.1 22.834 a 22.974). La mutante del gen ATP18 no afecta el crecimiento en medio no fermentable, lo que sugiere que no es esencial para la función de la enzima de levadura (Paumard et al., 2000). Estudios previos con entrecruzadores demostraron la interacción de los heterodímeros i+i, i+f, i+d, así como la interacción de los subcomplejos i+6+f e i+6+d (Figura 13). El homodímero de la subunidad i se ha demostrado participa en la oligomerización de la enzima de levadura (Paumard et al., 2000).

Representación esquemática del modelo de topología de la subunidad i. El modelo está basado en los ensayos empleando agentes entrecruzadores, en donde se representa esquemáticamente a las proteínas cercanas a la subunidad i. La numeración de la subunidad 6 inicia en la Ser1. Se muestra el primer segmento que atraviesa la membrana y el primer bucle situado del lado de la matriz de la subunidad 6. Los círculos negros representan los residuos de lisina sustituidos por cisteínas en la subunidad i. Otros residuos de lisina se muestran como círculos con líneas. 4D54C significa el cambio del residuo Asp54 por Cis de la subunidad 4. P = lado positivo, N = lado negativo. Modificada de Paumard et al., 2000.
Recientemente dos nuevas proteínas, llamadas k (descrita en S. cerevisiae) y l (reportada en Y. lipolytica, Pichia pastoris, Pichia angusta y S. cerevisiae), se han asociado al dominio FO. Análisis bioinformáticos sugieren que ambas presentan su extremo carboxilo expuesto al espacio intermembranal y su función putativa está relacionada al ensamblaje del complejo enzimático (Arnold et al., 1998; Liu et al., 2015). En las levaduras, las subunidad k e i estabilizan al dímero de la F1FO-ATP sintasa (Wagner et al., 2010).
Factor B y proteína TMEM70El factor b de bovino no tiene homólogos en procariontes ni una contraparte en la levadura, parece unirse del lado de la matriz de la membrana interna mitocondrial y no parece tener una función reguladora. Además, de la vía de translocación de protones clásica descrita para el dominio FO, se ha propuesto que la región membranal de los mamíferos podría albergar una segunda vía de translocación de los protones, formada a partir del ensamblaje de las subunidades e, f, g y A6L, así como por los segmentos transmembranales del acarreador ADP/ATP. También, se ha sugerido que este factor puede ocluir esta segunda vía de translocación de protones favoreciendo así la actividad de la ATP sintasa. Sin embargo, no se sabe con detalle su función dentro del complejo FO (Jonckheere et al., 2012).
La proteína transmembrana 70 (TMEM70) de bovino tiene una masa molecular aproximada de 21 kDa. Análisis de fracciones submitocondriales han demostrado que TMEM70 se asocia con la membrana interna mitocondrial. Se ha mostrado que esta proteína se requiere para mantener los niveles normales de expresión y actividad del complejo V. Aunque no se ha determinado una interacción directa con el complejo enzimático, se ha propuesto una unión transitoria con la enzima. TMEM70 es una proteína poco abundante, y al igual que los factores de ensamblaje ATP11 y ATP12, se ha sugerido que participa en la biogénesis del complejo V, no obstante se requieren más estudios para aclarar su función (Jonckheere et al., 2012).
Proteínas MLQ y AGPRecientemente se han descrito dos nuevas proteínas asociadas al complejo enzimático de bovino. La primera de ellas se conoce como proteolípido mitocondrial de 6.8 kDa (6.8PL) o proteína MLQ debido a que se obtuvo a partir de membranas mitocondriales empleando disolventes orgánicos (cloroformo/metanol). La segunda es una subunidad llamada AGP y se ha descrito como una proteína asociada a la diabetes en tejidos sensibles a insulina (DAPIT, por sus siglas en inglés). Se ha determinado que estas subunidades no afectan la actividad del complejo V. Empleando servidores bioinformáticos se predice un cruce transmembranal para cada una de ellas, dos sitios de fosforilación posibles para AGP y uno para la proteína MLQ. Al buscar en la base de datos del genoma de levadura homólogos para estas subunidades se observó que sus secuencias tenían una baja identidad (13%) y similitud (16%). Respecto a su función, aún no se conoce con precisión. No obstante, estudios realizados en tejidos de rata en un modelo de diabetes, inducido al dañar las células β pancreáticas debido a la administración del fármaco estreptozotocina, sugieren que podría participar en el metabolismo de la glucosa y/o la fosforilación oxidativa. En este sentido, se ha diseñado RNA de interferencia para evaluar modificaciones morfológicas de la membrana interna mitocondrial o cambios en las formas oligómericas de la ATP sintasa, sin embargo, no se han obtenido resultados significativos (Meyer et al., 2007).
Organización estructural del dominio membranalInicialmente se pensó que las subunidades e y g estabilizaban al dímero de la ATPasa, mientras que la subunidad b favorecía la oligomerización de la enzima. Sin embargo, los heterodímeros e+b y g+b obtenidos por estudios de entrecruzamiento, proponían que estas subunidades estabilizaban la interfaz monómero-monómero en la ATP sintasa dimérica. De la misma forma, el homodímero i+i, además de los productos de entrecruzamiento como e+b, g+b, h+b y e+i+e sugerían que al menos cinco subunidades cercanas, e, g, i, h y b, formaban parte de la interfaz monomérica del complejo enzimático. Tomando en cuenta estos resultados, así como el elevado número de alfa hélices membranales (aunque esta predicción varía de acuerdo con el algoritmo empleado, con la excepción de la subunidad h que no presenta una hélice membranal) refuerzan la idea de que las interacciones de las proteínas del sector FO participan en la oligomerización de la enzima (Wittig & Schägger, 2008). En las levaduras, la subunidad h se asocia al sector F1 y no está involucrada en la dimerización (Arselin et al., 1996; Couoh-Cardel et al., 2010).
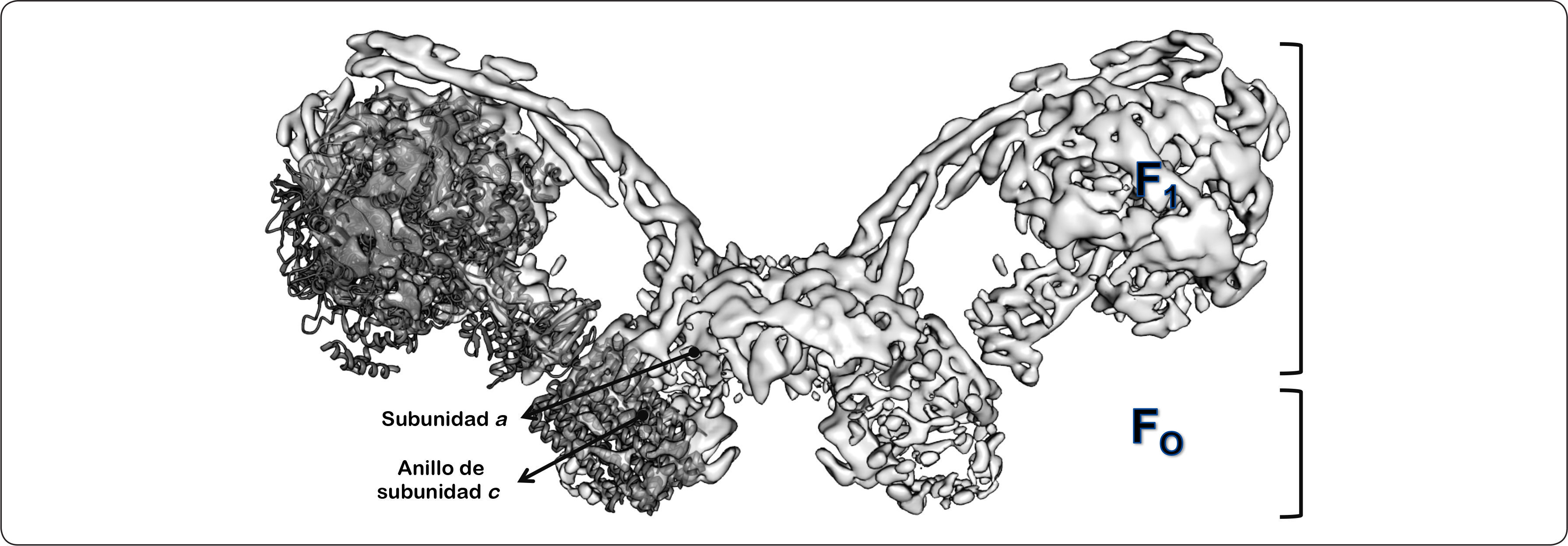
Como se ha expuesto, la alta hidrofobicidad de las subunidades del dominio membranal ha dificultado su caracterización. Actualmente, se han realizado esfuerzos experimentales para determinar la estructura de la subunidades del sector FO de la ATPasa mitocondrial de Yarrowia lipolytica. Empleando una combinación de técnicas de alta resolución estructural, como la crío-microscopía electrónica y la cristalografía de rayos X, se ha determinado la estructura completa del dímero de la levadura con una resolución general de 7.8Å, de 6.2Å para el dominio FO del dímero de la enzima y de 6.9Å para el sector F1 del monómero del complejo enzimático. El mapa de densidad electrónica obtenido experimentalmente permitió identificar a la mayoría de las subunidades del dominio FO (Figura 14) (Hahn et al., 2016).
. Adicionalmente, a manera de comparación sobre la densidad electrónica del mismo se muestra la estructura cristalográfica del sub-complejo F1-c10 de Y. lipolytica (3.5Å de resolución, no. de acceso PDB 5FL7). Asimismo, se indica la región correspondiente a los dominios extrínseco (F1) e intrínseco de membrana (FO) del complejo dimérico de la enzima de la levadura, así como las subunidades a y el anillo de subunidades de c. Modificado de Hahn et al., 2016.")
Modelo estructural de la F1FO-ATP sintasa dimérica de Yarrowia lipolytica.Vista lateral del mapa de densidad electrónica obtenido por crío-microscopía electrónica del dímero de la ATPasa de Y. lipolytica representado como un diagrama de superficie (7.8Å de resolución, no. de acceso EMD 8151). Adicionalmente, a manera de comparación sobre la densidad electrónica del mismo se muestra la estructura cristalográfica del sub-complejo F1-c10 de Y. lipolytica (3.5Å de resolución, no. de acceso PDB 5FL7). Asimismo, se indica la región correspondiente a los dominios extrínseco (F1) e intrínseco de membrana (FO) del complejo dimérico de la enzima de la levadura, así como las subunidades a y el anillo de subunidades de c. Modificado de Hahn et al., 2016.
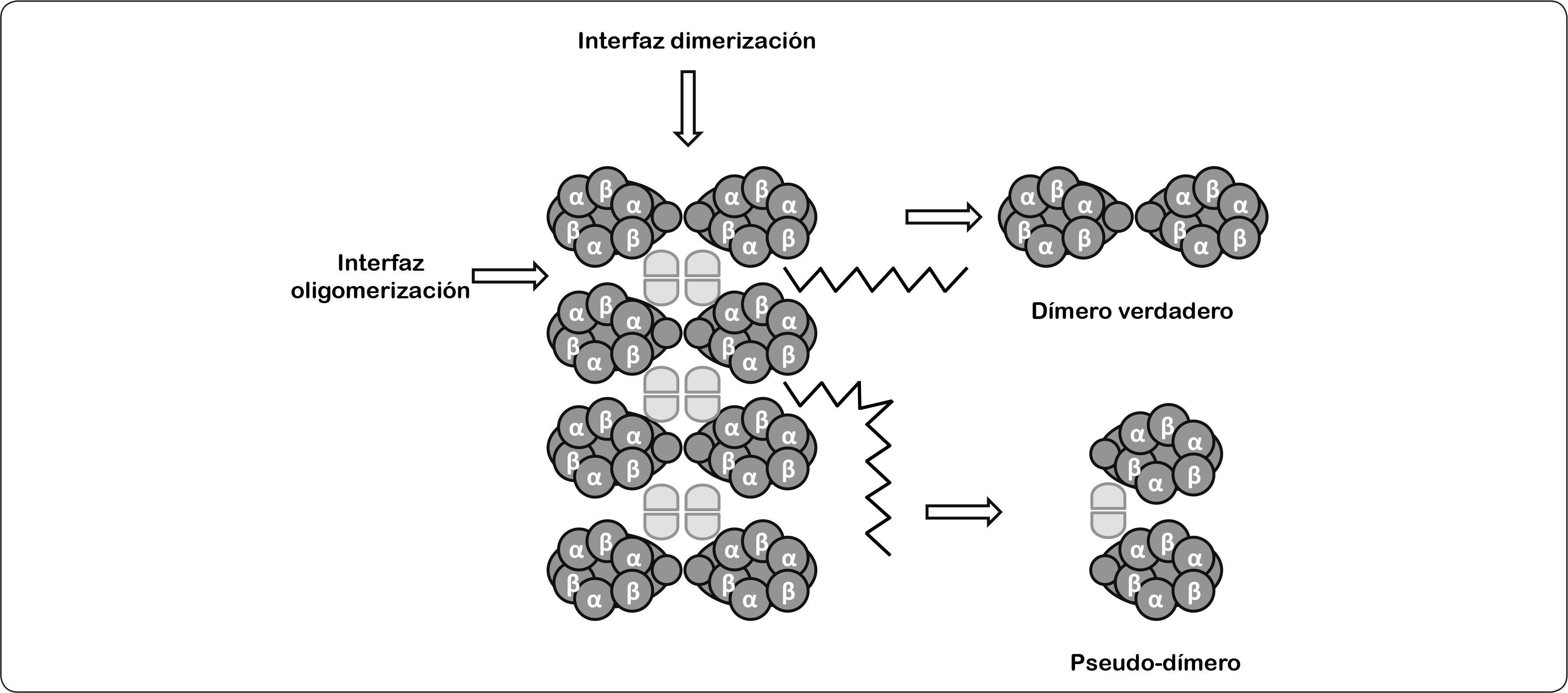
La organización supramolecular de la ATP sintasa a lo largo del borde de las crestas mitocondriales se observó por primera vez en el protozoario ciliado Paramecium multinucleatum; el análisis de las imágenes de microscopía de crío-fractura permitieron establecer una relación entre los dímeros de la enzima con la formación de crestas mitocondriales (Paumard et al., 2002). Recientemente, estas formaciones diméricas se han descrito por microscopía de fuerza atómica y microscopía electrónica en las ATPasas de rata, bovino, levadura, papa y hongos. A la fecha, la existencia de hileras de dímeros de la ATP sintasa mitocondrial es aceptada. Estudios realizados con agentes entrecruzadores han caracterizado una interfaz de dimerización (subunidades 6, 4, i, e) y otra de oligomerización (subunidades i, e, g), a través de las cuales se dan los contactos entre los dímeros de la enzima (Figura 15) (Habersetzer et al., 2013). En este sentido, se encontró que los monómeros del bovino, la levadura y las algas clorofíceas formaban dos ángulos distintos de asociación lo que sugería la existencia de dos tipos de estructuras, una llamada “dímero verdadero” con un ángulo de 70-90° y otra conocida como “pseudo-dímero” cuyo ángulo es menor a 40°, dando lugar a la formación de crestas mitocondriales lamelares o tubulares respectivamente. Por otra parte, se ha propuesto que estos dímeros podrían ser el resultado de la ruptura de los oligómeros de la ATPasa de las crestas mitocondriales (Figura 15) (Dudkina et al., 2006).
. Así como las superficies de contacto que forman los monómeros de la enzima (representados como elipses horizontales), y dentro de éstos las subunidades α/β. También, se indica la interfaz de dimerización y oligomerización (señalado con una flecha), respectivamente. Modificado de Dudkina et al., 2006.")
Modelo de la organización de las estructuras oligoméricas de la ATP sintasa. En este modelo se representa de manera esquemática la disposición de los multímeros de la ATPasa. Se muestran los dos tipos de formas diméricas que existen (dímero verdadero y pseudo-dímero). Así como las superficies de contacto que forman los monómeros de la enzima (representados como elipses horizontales), y dentro de éstos las subunidades α/β. También, se indica la interfaz de dimerización y oligomerización (señalado con una flecha), respectivamente. Modificado de Dudkina et al., 2006.
En comparación con la estructura de la ATP sintasa de los mamíferos o de la levadura, que han sido muy estudiadas, poco se sabe de la composición estructural de la ATP sintasa de las plantas (Cano-Estrada & González-Halphen, 2011). En éstas, aún menos subunidades del sector FO han sido caracterizadas, y solamente se han reportado genes que codifican para las subunidades a y c. No obstante, en el genoma mitocondrial de Arabidopsis thaliana se han encontrado los genes orfB y orf25 que codifican para proteínas asociadas al dominio membranal de la ATP sintasa. El gen orf25 codifica para una proteína denominada FAd. Sin embargo, los productos proteícos de estos genes no tienen homólogos claros en bovino o levadura y su función es aún desconocida (Heazlewood et al., 2003).
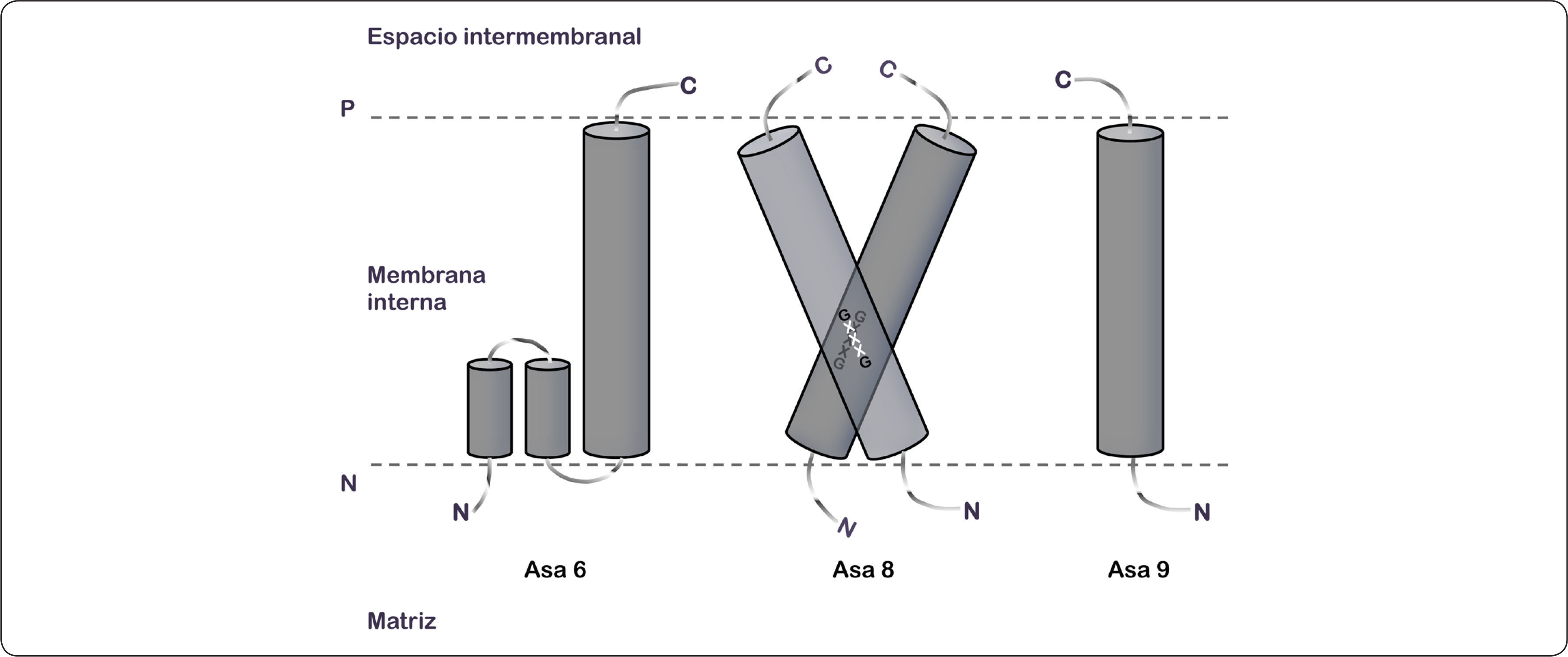
En contraste con otros complejos enzimáticos, el brazo periférico y el dominio membranal de las ATPasas de las algas Chlamydomonas reinhardtii y Polytomella sp. están compuestos por nueve polipéptidos atípicos (Asa1-9) (Vázquez-Acevedo et al., 2006). El análisis de imágenes de microscopía electrónica de las secciones transversales de la membrana reveló seis hélices transmembranales (probablemente siete) (Allegretti et al., 2015), adicionales a las correspondientes de las subunidades a y c, que en principio podrían atribuirse a las subunidades Asa6, Asa8 y Asa9. Sin embargo, la resolución de la imagen estructural obtenida en este dominio membranal dificulta la correcta asignación para cada una de las subunidades que lo componen.
La subunidad Asa6 posee 151 aminoácidos (13.1 kDa), su extremo amino se encuentra orientado hacia la matriz mitocondrial y su dominio carboxilo está ubicado en el espacio intermembranal. Algunos servidores de predicción de estructura secundaria para proteínas de membrana sugieren dos cruces transmembranales, mientras que otros algoritmos predicen una hélice re-entrante y un cruce membranal. Asimismo, Asa8 se compone de 89 aminoácidos (9.9 kDa), presenta un cruce transmembranal y una topología de membrana N-adentro C-fuera, deducido a partir de análisis bioinformáticos. Debido a que la subunidad Asa8 contiene un dominio GxxxG (Gly59-Gly63) podría formar homodímeros y de esta forma participar en la dimerización de la enzima (Vázquez-Acevedo et al., 2016), ya que la presencia de estos dominios en las subunidades e y g de levadura desempeñan un papel importante en la formación de estructuras diméricas del complejo enzimático (Arselin et al., 2003; Arselin et al., 2004; Bustos & Velours, 2005). Por último, Asa9 está formada por 97 aminoácidos y tiene una masa molecular estimada de 11 kDa. Al igual que las subunidades anteriores, este polipéptido presenta la misma topología de membrana y por lo menos un cruce transmembranal (Figura 16) (Vázquez-Acevedo et al., 2016).
. Los modelos predichos para Asa6 varían con los diferentes programas que se utilizaron: un TMS, dos TMS, o un modelo con un TMS y una hélice re-entrante (representado). TMS = Segmento transmembranal. P = lado positivo, N = lado negativo. Modificado de Vázquez-Acevedo et al., 2016.")
Modelos basados en la hidrofobicidad de las subunidades individuales que participan en el dominio de dimerización de membrana. Varios algoritmos bioinformáticos predicen las subunidades Asa8 y Asa9 con un solo TMS. El motivo GxxxG presente en la subunidad Asa8 podría llevar a su dimerización (representado). Los modelos predichos para Asa6 varían con los diferentes programas que se utilizaron: un TMS, dos TMS, o un modelo con un TMS y una hélice re-entrante (representado). TMS = Segmento transmembranal. P = lado positivo, N = lado negativo. Modificado de Vázquez-Acevedo et al., 2016.
Por otra parte, en protozoarios y ciliados al igual que en las algas clorofíceas, se han encontrado subunidades novedosas sin homólogos en otros organismos. Para el primer caso, Trypanosoma brucei, se identificaron 22 proteínas, de las cuales cinco están relacionadas a la región F1 y tres al sector FO, mientras que las 14 restantes se asignaron como proteínas putativas (Zíková et al., 2009). En el segundo caso, Tetrahymena thermophila, se encontraron siete ortólogos para las subunidades clásicas de ATPasa, con excepción de la subunidad c. Además de las 13 subunidades novedosas, del total de proteínas encontradas, se descubrió que la proteína Ymf66 tiene ochos segmentos membranales y podría sustituir a la subunidad a en este complejo enzimático (Balabaskaran et al., 2010).
CONCLUSIONESEl complejo enzimático F1FO-ATP sintasa es un nano-motor rotatorio capaz de sintetizar ATP aprovechando la fuerza protón motriz. Su sector F1 cataliza la síntesis de ATP, mientras que el sector FO lleva a cabo la translocación de iones y proporciona un estator para la acción giratoria del complejo (Heazlewood et al., 2003).
La enzima de la mayoría de los organismos eucariontes, desde las levaduras hasta las plantas y los mamíferos, cuentan con las mismas subunidades, aunque poco conservadas a nivel de secuencia pero más conservadas a nivel de estructura, lo cual indica que estas subunidades comparten la misma función dentro del complejo (Vázquez-Acevedo et al., 2016).
Las principales diferencias estructurales de la ATP sintasa de los diferentes organismos se encuentran en la composición polipeptídica del brazo periférico y en el dominio membranal; estas diferencias en algunos casos son dependientes de la especie. Por ejemplo, en las algas clorofíceas, los ciliados y los protozoarios existen subunidades atípicas que estabilizan el complejo enzimático y optimizan el metabolismo energético celular en estos organismos.
El estudio de la composición de las subunidades del complejo FO se ha abordado mediante diversas estrategias experimentales: análisis de la composición polipeptídica mediante geles en condiciones desnaturalizantes, fraccionamiento de las subunidades mediante técnicas de purificación y análisis por espectrometría de masas, diseño de mutantes para los distintos genes que codifican para las proteínas del complejo enzimático y empleo de agentes entrecruzadores, entre otros. Sin embargo, las dificultades experimentales que resultan de la alta hidrofobicidad de las proteínas, por ejemplo las subunidades a, c y A6L, dificultan su identificación aunque estén presentes en cantidades equimolares. Por tal motivo, no se puede excluir la posibilidad que una o más proteínas no sean detectadas o se consideren contaminantes. Por lo tanto, es difícil asegurar que no existan subunidades adicionales en los dominios membranales por descubrir. No obstante, gracias al desarrollo de nuevas técnicas para el análisis estructural de proteínas, como la crío-microscopía electrónica, se puede estudiar con mayor detalle la composición de las subunidades intrínsecas de la membrana.
AGRADECIMIENTOSAgradecemos a la Dra. Marietta Tuena Sangri (Instituto de Fisiología Celular, UNAM) y al Dr. Óscar Flores Herrera (Facultad de Medicina, UNAM) por sus valiosos comentarios críticos al presente manuscrito. Agradecemos el apoyo técnico de la QBP. Miriam Vázquez-Acevedo y el apoyo de los siguientes donativos: CONACYT, México (239219); Cooperación Bilateral CONACYT-FNRS, México-Bélgica (245486) y a la DGAPA-UNAM, México (IN203311). Lorenzo Sánchez-Vásquez es estudiante del Programa de Doctorado en Ciencias Biomédicas (PDCB) de la Universidad Nacional Autónoma de México (UNAM) y recibió la beca 128110 del CONACYT.