


El angiomiolipoma (AML) es un tumor benigno de origen mesenquimal, compuesto por tejido adiposo maduro, vasos sanguíneos aberrantes y músculo liso, con una prevalencia aproximada del 1-3% de todas las masas renales. Se han descrito presentaciones aisladas y relacionadas esporádicamente con Linfangioleiomiomatosis pulmonar y/o en el contexto del síndrome de esclerosis Tuberosa, siendo estos últimos 2 contextos, más difíciles en cuanto a diagnóstico y manejo de intención curativa. Presentamos el caso de una paciente diagnosticada con Angiomiolipoma Renal Gigante y Linfangioleiomiomatosis pulmonar esporádica no filiada, diagnosticada y manejada con nefrectomía izquierda previa embolización selectiva arterial, en la Fundación Cardiovascular de Colombia.
ConclusionEl Angiomiolipoma Renal es una enfermedad de baja prevalencia, de diagnóstico incidental en su mayoría, sin que requiera de intervención más que seguimiento por el urólogo. Sin embargo, cuando nos enfrentamos a pacientes con grandes masas tumorales y/o múltiples, sumado a manifestaciones clínicas, habrá de pensar entre otros, a asociación con Linfangioleiomiomatosis Pulmonar y/o síndrome de esclerosis tuberosa, entidades que constitu yen un reto desde el diagnostico hasta el tratamiento por un grupo multidisciplinario, en un intento por mejorar la calidad de vida de las afectadas.
© 2013 Sociedad Colombiana de Urología. Publicado por Elsevier España, S.L. Todos los derechos reservados.
Angiomyolipoma (AML) is a benign mesenchymal tumor composed of mature adipose tissue, aberrant blood vessels and smooth muscle, with a prevalence of approximately 1-3% of all renal masses. It has been reported as an isolated presentation, as sporadic pulmonary lymphangioleiomyomatosis and / or in the context of tuberous sclerosis syndrome, with the last 2 contexts being more difficult in terms of diagnosis and management of curative intent. The case is presented of a patient diagnosed with giant renal angiomyolipoma and pulmonary lymphangioleiomyomatosis, diagnosed and managed with left nephrectomy prior to arterial embolization in the Fundación Cardiovascular de Colombia.
ConclusionRenal angiomyolipoma is a disease of low prevalence, of incidental diagnosis in most cases, and does not require intervention but rather follow-up by the urologist. However, when faced with patients with large tumors and / or multiple masses, coupled with clinical manifestations, an association with pulmonary lymphangioleiomyomatosis and / or tuberous sclerosis should be kept in mind, as they are entities that are a challenge from diagnosis to treatment by a multidisciplinary team, in order to improve the quality of life of affected patients.
© 2013 Sociedad Colombiana de Urología. Published by Elsevier España, S.L. All rights reserved.
El Angiomiolipoma (AML) es un tumor benigno de origen mesenquimal, compuesto por tejido adiposo maduro, vasos sanguíneos aberrantes y músculo liso, con una prevalencia aproximada del 1-3% de todas las masas renales, entidad que ha mostrado un incremento progresivo en su diagnóstico, gracias al avance en estudios de imágenes, que son tomados rutinariamente o por otras indicaciones. Aproximadamente el 80% son esporádicos y asociados a la mutación del gen TSC2, mientras que hasta un 20% están asociados con el síndrome de esclerosis tuberosa (TSC), relacionado con el gen TSC1 y de forma esporádica con Linfangioleiomiomatosis1–4 (LAM). En sentido contrario, entre el 55 y el 80% de los pacientes con TSC y TSC-LAM, se les documentará AML2,3,5,6, Aunque en términos generales es considerada una neoplasia benigna, se han descrito casos raros de extensión a vena renal y cava inferior, además de infiltración regional a ganglios, e incluso colon1.
Debido a que se trata de una patología poco frecuente, describimos el caso de una paciente adulto joven, quien fue diagnosticada con Angiomiolipoma Renal Gigante y Linfangioleiomiomatosis Pulmonar, y manejada en la Fundación Cardiovascular de Colombia, seccional Floridablanca, por el grupo interdisciplinario de urología, neumología y genética, en el primer trimestre de 2013.
Reporte de casoPaciente femenina adulto medio de 48 años, odontóloga y docente universitaria, quien ingresa a urgencias por cuadro clínico caracterizado por dolor torácico opresivo durante tiempo no establecido, mientras realizaba actividad física, además de un episodio de sincope, sin otra sintomatología asociada. Como antecedentes patológicos dislipidemia y Trastorno Depresivo Mayor (TDM), manejados con atorvastatina y antidepresivos respectivamente. A la revisión por sistemas manifiesta disnea progresiva hasta hacerse de pequeños esfuerzos, de aproximadamente tres meses de evolución, acompañada de dolor torácico ante el esfuerzo físico. Al examen físico de ingreso destacaba buen estado general, sobrepeso. La auscultación cardíaca evidenciaba soplo pan-sistólico sin irradiación, acompañado de S2 reforzado en focos de la base. Al examen abdominal, no se evidencia visceromegalias, masas o adenopatías, sin embargo puño-percusión izquierda positiva. Se inician estudios de electrocardiografía y ecocardiografía que mostraron, respectivamente, taquicardia sinusal, con signos de sobrecarga ventricular derecha, signos de hipertensión pulmonar grave con dilatación y disfunción del ventrículo derecho.
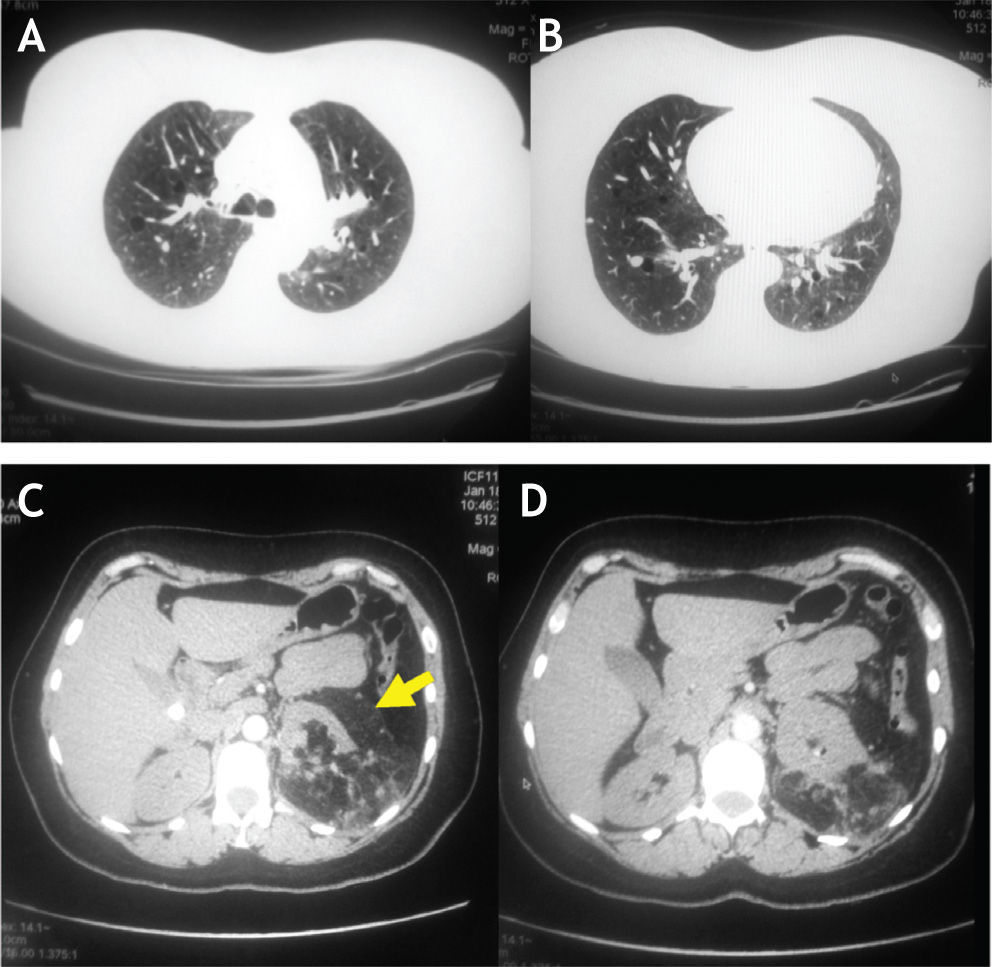
La tomografía axial computada (TAC) de tórax mostró alteraciones quísticas bilaterales en parénquima pulmonar, nódulo subpleural de pequeño tamaño en segmento basal lateral del LID de etiología a determinar y lesión tipo masa de aspecto sospechoso de angiomiolipoma en polo renal izquierdo (figs. 1A y B, flechas pequeñas; figs. 1C y D, flecha grande). Por lo anterior, se completa estudio con TAC contrastado de abdomen el cual muestra masa heterogénea, que compromete el riñón izquierdo, de 15 × 15 cm, de diferentes densidades entre 10 y 50 UH.
: Tomografía axial de tórax. C y D (abajo): Tomografía axial de abdomen.")
Se decide ingreso hospitalario, se solicita hemodinámica pulmonar para confirmación y estadificación de hipertensión arterial pulmonar con test vasodilatador pulmonar, filiación funcional respiratoria, evaluación de intercambio de gases, cribado de neoplasia mediante marcadores tumorales, y se consideró toma de biopsia de pulmón para estudio histopatológico. Sin embargo, el procedimiento fue pospuesto atendiendo al alto riesgo intra y postoperatorio secundario a hipertensión pulmonar. Los resultados mostraron una alteración de tipo restrictivo de moderada intensidad, sin deterioro de la transferencia del monóxido de carbono.
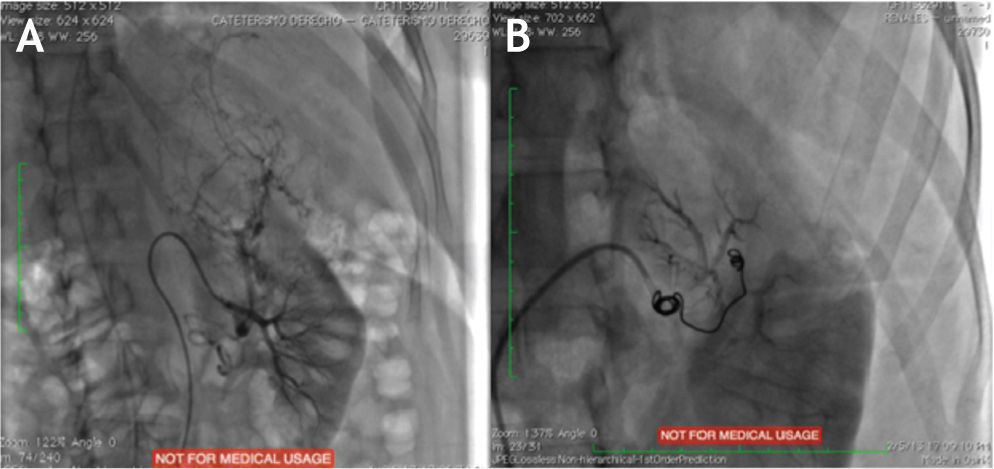
Es valorada por el servicio de Urología, quienes consideraron una vez revisada la clínica y examinado a la paciente, y de acuerdo con las siguientes condiciones clínicas: 1. Gran volumen tumoral, lo que aumenta el riesgo de ruptura y sangrado espontaneo secundario por un lado; masa que a su vez condiciona restricción mecánica extrínseca de la ventilación, influyendo en la fisiopatología de los síntomas pulmonares de la paciente, y 2. Reporte de algunos casos de comportamiento agresivo y/o atípico7, era candidata a nefrectomía izquierda, 24 horas después de embolización selectiva de arteria lobar superior renal, en búsqueda de disminución tanto de volumen tumoral y riesgo de sangrado intraoperatorio1–3,6,8 (fig. 2). Aunque si bien la Linfangioleiomiomatosis pulmonar es una patología que no presenta según los reportes de la literatura un pronóstico favorable a mediano y corto plazo, al tratarse de una paciente joven, previamente sana, a la globalmente no se le puede estimar una expectativa de vida con un diagnostico reciente, además del alto riesgo de sangrado secundario a el Angiomiolipoma renal gigante y muerte inminente secundario a este, se decidió nefrectomía izquierda, en búsqueda de un manejo integral y mejorar su calidad de vida.

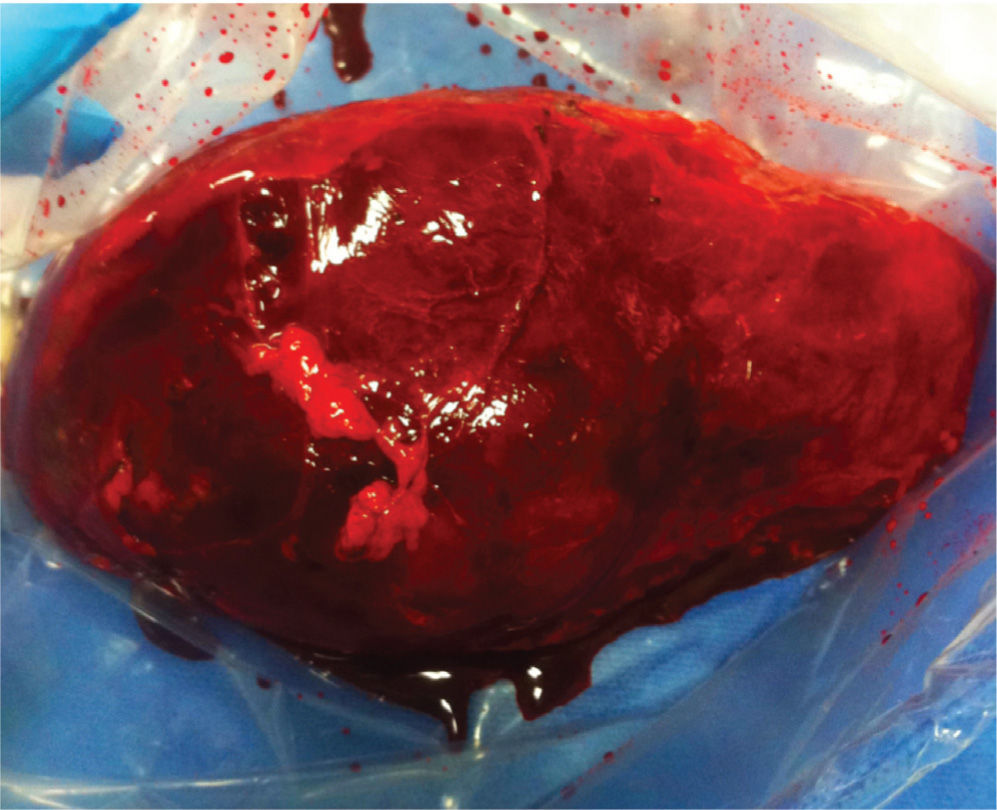
Se realiza nefrectomía por técnica abierta, con incisión de hemichevron amplia izquierda, encontrándose masa gigante con aspecto eminentemente vascular, de consistencia blanda, adherida a bazo páncreas y Colón. En el post-operatorio se desarrolló de derrame pleural bilateral, de características exudativas, sin gérmenes, asociado a síntomas y signos de refractariedad ante el tratamiento vasodilatador pulmonar, con posterior insuficiencia respiratoria grave y atelectasia lobar izquierda, por lo cual se inicia ventilación mecánica no-invasiva. Adicionalmente la paciente cursa con imágenes acino-alveolares algodonosas, por lo que se decide iniciar tratamiento antibiótico con piperacilina tazobactam. El resultado de patología confirma que se trata de un Angiomiolipoma (fig. 3).

Presente síndrome febril agudo en el 29 día postoperatorio, por lo que se toman hemocultivos y TAC de abdomen contrastado que muestra pequeña colección de 50 cc en región subfrénica izquierda, se lleva a revisión vía abierta por cirugía general, tomándose cultivos de los hallazgos, se inicia antibioticoterapia para germen nosocomial con ertapenem por 7 días, siendo el reporte final de hemocultivos y cultivo de secreción negativos. Posteriormente cursa con síndrome de dificultad respiratoria aguda, secundaria a derrames pleurales bilaterales importantes, requiriendo monitoreo en unidad de cuidado intensivo, realizándose toracentesis izquierda evacuadora de aproximadamente 500 ml, sin mejoría clínica, con acentuación de disnea, asociado a hipotensión, sospechándose sangrado activo a nivel torácico y/o abdominal, decidiéndose realización de toracotomía cerrada izquierda, con drenaje de 300cc de líquido hemático, sin respuesta, con fallecimiento por colapso circulatorio por disfunción ventricular severa secundaria a hipertensión pulmonar severa descompensada. Aunque por dificultades administrativas para lograr autorización en búsqueda de la confirmación por inmunohistoquímica para HMB-45, la paciente cumplía con los criterios clínicos y por imágenes de Linfangioleiomiomatosis pulmonar.
DiscusiónEl angiomiolipoma renal es un tumor de origen mesenquimal, el cual en la mayoría de los casos presenta un curso benigno, que como se mencionó previamente puede estar asociado a el síndrome de esclerosis tuberosa y/o asociarse de forma esporádica con Linfangioleiomiomatosis pulmonar, como en el caso de nuestra paciente1–3,5,6. Dentro del componente histológico, existen dos variantes: Típica y epitelioide, la segunda de estas con una mayor tasa de mitosis, pudiéndose encontrar recaída tumoral luego de su resección, mostrando por tanto un comportamiento más agresivo e incluso llegar a simular un carcinoma de células claras, diferenciándose de este, entre otros criterios, por su positividad para para el anticuerpo monoclonal derivado de los melanocitos HMB-45 2.
Dentro de las imágenes diagnósticas la Tomografía Axial Computada (TAC) es el estudio más ampliamente utilizado, siendo una herramienta útil para identificación y diagnóstico diferencial de masas que comprometen al riñón, dejando estudios invasivos como la biopsia percutánea para casos donde persisten dudas sobre el diagnostico1, procedimiento que puede aumentar el riesgo de sangrado. La mayoría de Angiomiolipomas, se diagnostican de forma incidental; sin embargo, existe un porcentaje cercano al 40%, en quienes se encuentra la triada clásica de dolor abdominal, hematuria y masa palpable4, y en menor proporción, debuta como abdomen agudo por hemorragia hemodinámicamente significativa por ruptura espontanea del tumor y/o de aneurismas intratumorales, cuadro que puede conllevar a shock hipovolémico e incluso la muerte, enmarcado dentro de las causas del síndrome de Wunderlich, requiriéndose intervención quirúrgica urgente para evitar dichos desenlaces2–4,6,9. En términos generales, se ha estimado que el riesgo de sangrado de un Angiomiolipoma pudiera llegar hasta un 50% según algunos reportes2.
Con cuanto al manejo, se han descrito seguimiento clínico, preservación de nefronas ya sea por embolización arterial selectiva (EAS) y nefrectomía parcial, o nefrectomía total, realizando una adecuada selección de pacientes candidatos a intervención como lo son: Pacientes sintomáticos, grandes masas tumorales o rápido crecimiento de las mismas, sangrado espontaneo del tumor o con alto riesgo de complicaciones, entre otros1–3,6. La embolización selectiva (EAS), conocida desde hace más de 20 años y cada vez más ampliamente utilizada como manejo definitivo, está indicada en pacientes con tumores múltiples, con alto riesgo quirúrgico y como primera medida en tumoraciones sangrantes, logrando una reducción significativa en el tamaño del mismo, con mínimas complicaciones que llegan al 10%, siendo el absceso renal la más frecuente3,8. Se ha descrito resangrado luego de una primera intervención hasta en un 20% de los pacientes. Ha mostrado resultados favorables, cuando se lleva a cabo previo a realización de nefrectomía total, al disminuir el riesgo de sangrado intraoperatorio, y de forma secundaria, las posibles complicaciones intra y postoperatorias6.
Dentro de los trastornos que se han correlacionado con el Angiomiolipoma, se encuentra la Linfangioleiomiomatosis, una condición descrita casi exclusivamente en mujeres jóvenes, en edad reproductiva, caracterizada por proliferación aberrante de células de musculo liso inmaduras (Células LAM) en varios órganos, siendo el principal blanco el pulmón con alteración de su parénquima, vasos linfáticos y sanguíneos, generando destrucción y remodelación quística, lo que conllevara a deterioro progresivo e irreversible sobre la capacidad funcional del mismo, que puede tener como resultado final la muerte de las afectadas por insuficiencia respiratoria5,10. Está enmarcada dentro de las enfermedades raras, dada su baja prevalencia, estimada tan solo en 1 caso por 1.000.000 de habitantes aproximadamente, su dificultad para llegar a un diagnóstico y ausencia de tratamiento curativo hasta el momento5. Suele ocurrir de forma esporádica (S-LAM), pero se ha asociado con el síndrome de esclerosis tuberosa (TSC) hasta en un 40%2,5,10,11. Alrededor del 60% de los pacientes con Linfangioleiomiomatosis (LAM) tendrán además AML1,2,5,10.
Tanto la variante epiteliode de AML como LAM, hacen parte del grupo de los PEComas (por sus siglas en inglés, Perivascular Epithelioid Cell), que hace referencia a tumores derivados de la proliferación clonal de las células epitelioides perivasculares1,7,10,11. Estas dos entidades comparten además características fenotípicas y genotípicas, entre las que incluyen la mutación en el gen TSC y tinción de marcadores específicos, vimectina, desmina y actina de musculo liso, además del anticuerpo HMB-452,5,11, hace que algunos autores hayan postulado que gracias a estas similitudes, y a la creciente evidencia que la lesión pulmonar en el TSC pudiera surgir de un Angiomiolipoma, que las estrategias que controlen efectivamente el crecimiento de células del Angiomiolipoma también pudieran llegar a detener la de células LAM2.
Dentro de las características fenotípicas de las células LAM, además de la expresión de algunos marcadores de diferenciación muscular, es la positividad frente a HMB-45 (Antígeno monoclonal frente a la glucoproteína pg100 de los premelanosomas), lo cual marca la diferencia frente a otros tipos celulares, haciendo de esta célula, un elemento con expresión fenotípica dual, dada la información antigénica de origen muscular liso y melanocítico, sugiriendo un origen en la cresta neural5,12. Los genes TCS1 y TCS 2 son quienes codifican las proteínas hamartina y tuberina, respectivamente, encargadas a su vez de inhibir el mTOR (mammalian target of rapamycin), regulador central del crecimiento celular a través de la síntesis proteica. Todo lo anterior, es relevante no solo para explicar la posible patogenia común entre LAM y TSC, sino también ser el punto de referencia hacia el futuro de terapias inmunosupresoras que apunten a controlar el crecimiento tumoral, al ejercer un efecto directo sobre el mTOR, como la rapamicina5.
Las manifestaciones clínicas dependerán de los órganos afectados, siendo las pulmonares las de mayor impacto y están directamente relacionadas con la proliferación de vasos linfáticos que atraviesan acúmulos de células LAM, que se ubican en la proximidad de los bronquios, vasos sanguíneos, pequeña vía aérea e incluso en la pleura, con obstrucción secundaria; De forma consecuente, cuando hay afectación de vasos sanguíneos llevara a hemorragia o hemoptisis, mientras que en la vía aérea será el atrapamiento aéreo, con disnea progresiva y tos secundaria. El Neumotórax, entidad de debut en algunas pacientes, sugiere la presencia de compromiso pleural, que cuenta con una tasa de recidiva entre el 61 y 81%5, requiriéndose una intervención temprana y agresiva con pleurodesis o pleurectomía, en un intento por disminuir estos porcentajes; a pesar de un tratamiento oportuno e indicado, hasta un 32% volverán a presentar otro evento5. En los vasos linfáticos, es característica la aparición de quilotórax por obstrucción y/o ruptura del conducto torácico, siendo difícil su abordaje terapéutico, pues dentro de su fisiopatología desencadena entre otras situaciones, algún grado de desnutrición e inmunosupresión, además que a pesar de las medidas encaminadas a disminuir la producción de quilo, la recidiva se convierte en la constante, por lo que en este caso también se requiere un manejo intensivo, dejando la ligadura del conducto torácico para casos graves5,10.
Matsui et al. propusieron en base a las características histológicas, una escala llamada LAM Histologic Score (LHS), de acuerdo al porcentaje de tejido pulmonar con componente quístico y proliferativo, en 3 grados de severidad pronostica, que se correlacionó con la supervivencia a 10 años de un 100% para LHS-1 (<25% de tejido comprometido), 74,4% para LHS-2 (25 al 50% de tejido) y 52,3% para LHS-3 (>50% de tejido pulmonar afectado), pudiéndose tener en cuenta como factor predictor de mortalidad y de indicación de trasplante de pulmón5,12,13.
De las manifestaciones extra-pulmonares, se ha documentado la aparición de linfadenopatías, linfangioleiomiomas y angiomiolipomas, los cuales generan en estadios avanzados de la enfermedad hasta en un 10% de los casos ascitis quilosa, que puede ocurrir por ruptura de un linfangioleiomioma y a su vez estar condicionada por la presencia concomitante de quilotorax5,10,12. Los linfangioleiomiomas que se producen entre un 16 al 21%12, son el resultado de la obstrucción linfática, más frecuentes en abdomen y pelvis, menos frecuentes en mediastino y cuello, manifestándose con dolor abdominal y edema de miembros inferiores, síntomas que empeoran con el paso del día por acumulo de líquido linfático en zonas de declive en la posición de bipedestación5.
La alteración abdominal más frecuente en pacientes con LAM, es sin duda el angiomiolipoma, dependiente del riñón en su mayoría, que suelen ser unilaterales, pequeños y restringidos al órgano cuando se presenta S-LAM (variante esporádica), mientras que en asociación con el síndrome de esclerosis tuberosa frecuentemente tienden a ser de mayor tamaño, múltiples, bilaterales, afectando varios órganos de forma simultánea y con mayor tendencia al sangrado2,5. TSC se ha relacionado con 3 fenotipos renales: Angiomiolipomas, quistes y carcinomas, siendo el primero el más frecuente2. Diferentes autores han encontrado exacerbación y progresión de los síntomas durante la gestación y/o con administración de terapia hormonal estrogénica, lo que llevo a pensar y posteriormente comprobar, la presencia de receptores para estrógenos y progestágenos en algunas de las células LAM, hecho ausente en las células musculares lisas normales. Cuando se detectan dichos receptores, las células LAM se muestran con un mayor tamaño, epitelioides y negativas para HMB-45; cuando por el contrario la expresión de receptores es negativa, son células más indiferenciadas y con mayor actividad proliferativa5. A pesar de este importante hallazgo, se ha comprobado la existencia de pacientes en gestación con diagnóstico de LAM que no exacerban su sintomatología, por lo cual aún no es claro el papel de la gestación con respecto a esta patología, requiriendo que siempre se individualice la conducta terapéutica de las afectadas5. Otras investigaciones han revelado que las células LAM tienen un potencial neoplásico, por tener una motilidad aumentada además de la capacidad para invadir la matriz colágena, detectándose células en orina, sangre y liquido quiloso, pudiendo explicar el poder de diseminación y de recidiva posterior en el tejido en el pulmón trasplantado5,12.
El diagnóstico definitivo se hará en base a la Tomografía Axial Computarizada de Alta Resolución de Tórax (TACAR), que muestra infiltraciones nodulares de pared fina intersticiales diseminadas homogéneamente en ambos campos pulmonares con zonas de parénquima intactos, asociado a cambios quísticos que rodean bronquios y estructuras vasculares linfohemáticas5,11,12, además de biopsia de parénquima pulmonar o ganglio linfático positivo para células LAM, que incluya estudio inmunohistoquímico de HMB-45, o con clínica sugestiva de TSC confirmada por angiolipomatosis o quilotórax5,11. La presencia de nódulos pulmonares no calcificados y angiolipomas, renales y/o hepáticos, son más prevalentes en TSC-LAM5. La espirometría suele mostrar un patrón obstructivo hasta en un 50% de las pacientes, de las que el 25% tendrá un test broncodilatador positivo y un descenso en la capacidad de difusión5. Existe una adecuada correlación entre la FEV1 calculada y la clasificación de LHS12. La capacidad pulmonar total, el volumen residual y su relación suele estar aumentada por hiperinsuflación y atrapamiento aéreo, además de una disminución de la tolerancia al ejercicio medida por prueba de esfuerzo5,12. Tanto la medición de FEV1 y la capacidad de difusión de monóxido de Carbono (DLCO) se convierten en buenos predictores de severidad y progresión de la enfermedad12.
El factor pronóstico más importante es el deterioro del intercambio gaseoso. Se estima una disminución en la FEV1 de 120 ml/año, según algunos autores5. Al parecer, el debut con neumotórax es más frecuente en mujeres jóvenes y se asocia a un mejor pronóstico, llegando a una supervivencia a 10años del 89%12, comparado con mujeres en quienes su síntoma cardinal es la disnea, en quienes la supervivencia es tan solo del 47% en el mismo periodo de tiempo5. Se ha estimado una tasa de supervivencia global de 60%11.
A pesar de los avances en el conocimiento de la fisiopatología, y de varios esquemas de manejo, en la actualidad no existe un tratamiento curativo. El uso de progesterona, tamoxifeno, análogos de las gonadotropinas e incluso ofoorectomía, herramientas utilizadas durante varios años, han sido cuestionadas pues ninguna ha mostrado un efecto significativo en la detención de la progresión de la enfermedad, y si se aumentan los efectos adversos5,12. Dentro del manejo integral, está descrito uso de broncodilatadores, aunque es poco el efecto, además de los corticoides inhalados o vía sistémica5. El trasplante de pulmón, se ha postulado como una opción en búsqueda de mejorar la supervivencia, el pronóstico a corto y mediano plazo, y mejorar la calidad de vida, para lo cual deben cumplirse requisitos estrictos en la selección de las pacientes. A pesar de las múltiples complicaciones peri y postoperatorias, diferentes publicaciones muestran una supervivencia de 80, 75 y 65% al uno, tres y cinco años respectivamente luego del trasplante5.
ConclusiónEl Angiomiolipoma Renal es una enfermedad de baja prevalencia, la cual ha venido siendo diagnosticada de forma creciente, en un porcentaje de casos de forma incidental gracias a estudios de imágenes por otras indicaciones. Dado que la mayoría de estos son de bajo volumen, únicos, y sin ninguna repercusión hemodinámica y/o manifestación clínica, se opta por manejo conservador y seguimiento clínico por el urólogo. Sin embargo, cuando nos enfrentamos a pacientes con grandes masas tumorales y/o múltiples, manifestaciones clínicas en otros sistemas como disnea, ascitis, neumotórax recurrente, o derrame pleural quiloso entre otros, habrá que pensar en una posible asociación con Linfangioleiomiomatosis Pulmonar, una entidad de baja prevalencia, curso clínico variable, la cual es de difícil manejo, que afecta predominantemente a mujeres en edad reproductiva, en búsqueda de un diagnóstico oportuno y manejo interdisciplinario para intentar mejorar la calidad de vida de las afectadas, dado que hasta el momento no se ha encontrado una terapia que logre un impacto significativo sobre el pronóstico a mediano y largo plazo sobre su historia natural dada la complejidad de la misma.
Nivel de evidenciaIII.
Conflicto de interesesLos autores declaran que no tienen conflicto de intereses.
Diseño del estudio: reporte de caso