



El síndrome de leiomiomatosis y cáncer renal hereditario, es un desorden de herencia autosómica dominante, que se caracteriza por la aparición de leiomiomas cutáneos, leiomiomas uterinos y un carcinoma renal papilar tipo 2 de mal pronóstico. Es causado por mutaciones de línea germinal en FH, un gen que codifica para la proteína fumarato hidratasa, la cual participa en el ciclo de Krebs.
Si bien es una enfermedad bastante rara, en el año 2016 Arenas et al., realizan el primer reporte de una familia colombiana con este síndrome y no se descarta que existan otras familias afectadas, por lo que mejorar el conocimiento sobre la enfermedad en la comunidad médica del país es crucial. En esta revisión, que partió de una búsqueda exhaustiva de literatura, se explica la fisiopatología y se proveen las principales pautas para el diagnóstico y tratamiento del síndrome.
Hereditary leiomyomatosis and renal cell cancer syndrome is an inherited autosomal dominant disorder that it is characterised by the appearance of cutaneous leiomyomas, uterine leiomyomas, and a type 2- papillary renal cell carcinoma, and with a poor prognosis. It is caused by germline mutations in FH, a gene that encodes the fumarate hydratase protein, and which take part in the Krebs cycle.
Although it is a very rare disease, Arenas et al., in 2016, presented the first report of a Colombian family with the syndrome. As its existence in other families cannot be ruled out, it is essential to improve the knowledge of the disease among the medical community of this country. In this review, which was based in an exhaustive search of literature, the pathophysiology is explained, and the main guidelines for the diagnosis and treatment of this syndrome are provided.
El síndrome de leiomiomatosis y cáncer renal hereditario (HLRCC por sus siglas en inglés) es una enfermedad bastante rara de la cual no existen por ahora datos de incidencia ni de prevalencia. Desde su primer reporte en el año 1973 por Reed et al.1, un poco más de 200 familias en el mundo han sido descritas en la literatura mundial, siendo estas en su mayoría procedentes de Norteamérica2,3, Reino Unido, Holanda y Finlandia2.
Se trata de una patología de herencia autosómica dominante que puede cursar con 3 manifestaciones clínicas principales como son: leiomiomas cutáneos, leiomiomas uterinos y cáncer renal papilar tipo 2, este último de mal pronóstico, inicio temprano y comportamiento agresivo4.
En este artículo se realiza una revisión de los aspectos más relevantes de esta enfermedad, a propósito del reporte por parte de nuestro grupo de investigación, de hasta donde sabemos, la primera familia colombiana con el síndrome HLRCC; en la que llamaba la atención la presencia a lo largo de 4 generaciones de individuos afectados por el cáncer renal papilar tipo 2, que fallecieron por esta causa a edades tempranas5.
En este sentido, se realizó una caracterización clínica y genética de 20 miembros de esta familia, y en 6 de ellos –incluyendo el probando, único afectado por el momento con el cáncer renal– se identificó una mutación nunca antes descrita en el gen FH denominada c.1349_1352delATGA, la cual genera un truncamiento de la proteína y por tanto se considera patogénica. En ninguno de los individuos afectados se evidenciaron leiomiomas cutáneos, que son la manifestación más común de la enfermedad; pero sí se documentaron quistes renales, algunos tumores poco comunes dentro del espectro del HLRCC y leiomiomas uterinos en las mujeres5.
Así las cosas, el objetivo fundamental de este artículo es hacer una aportación al conocimiento de la enfermedad en nuestro país, en un esfuerzo por mejorar el manejo de quienes padecen este síndrome, el cual creemos puede estar siendo subdiagnosticado.
Método de búsquedaSe hizo una revisión de tipo narrativo. La búsqueda de literatura fue realizada en las base de datos de Pubmed, Science direct y Springer Link utilizando como palabras clave: HLRCC, fumarato hidratasa, FH, diagnóstico y tratamiento. Los límites de la búsqueda se establecieron entre 1970 y 2016 y se excluyeron artículos en idiomas distintos al inglés.
GeneralidadesEl síndrome HLRCC es una enfermedad de predisposición a cáncer, cuya caracterización clínica y molecular se ha dado en un periodo relativamente reciente; no fue sino hasta 2001 que se estableció con total certeza que se trataba de una enfermedad con una herencia dominante, en la que algunos afectados podían presentar además de los leiomiomas cutáneos y uterinos, un raro tipo de cáncer renal, el papilar tipo 26. En el año 2002, Tomlinson et al.7 descubren que se produce por mutaciones de línea germinal en el gen FH (1q42.3-43), con lo que se empieza a dilucidar su fisiopatología.
Aspectos genéticos y moleculares del síndrome de leiomiomatosis y cáncer renal hereditarioEl síndrome HLRCC es generado por mutaciones de línea germinal en el gen FH (1q42.3-43), el cual posee 10 exones y codifica para la proteína fumarato hidratasa, de 510 aminoácidos7.
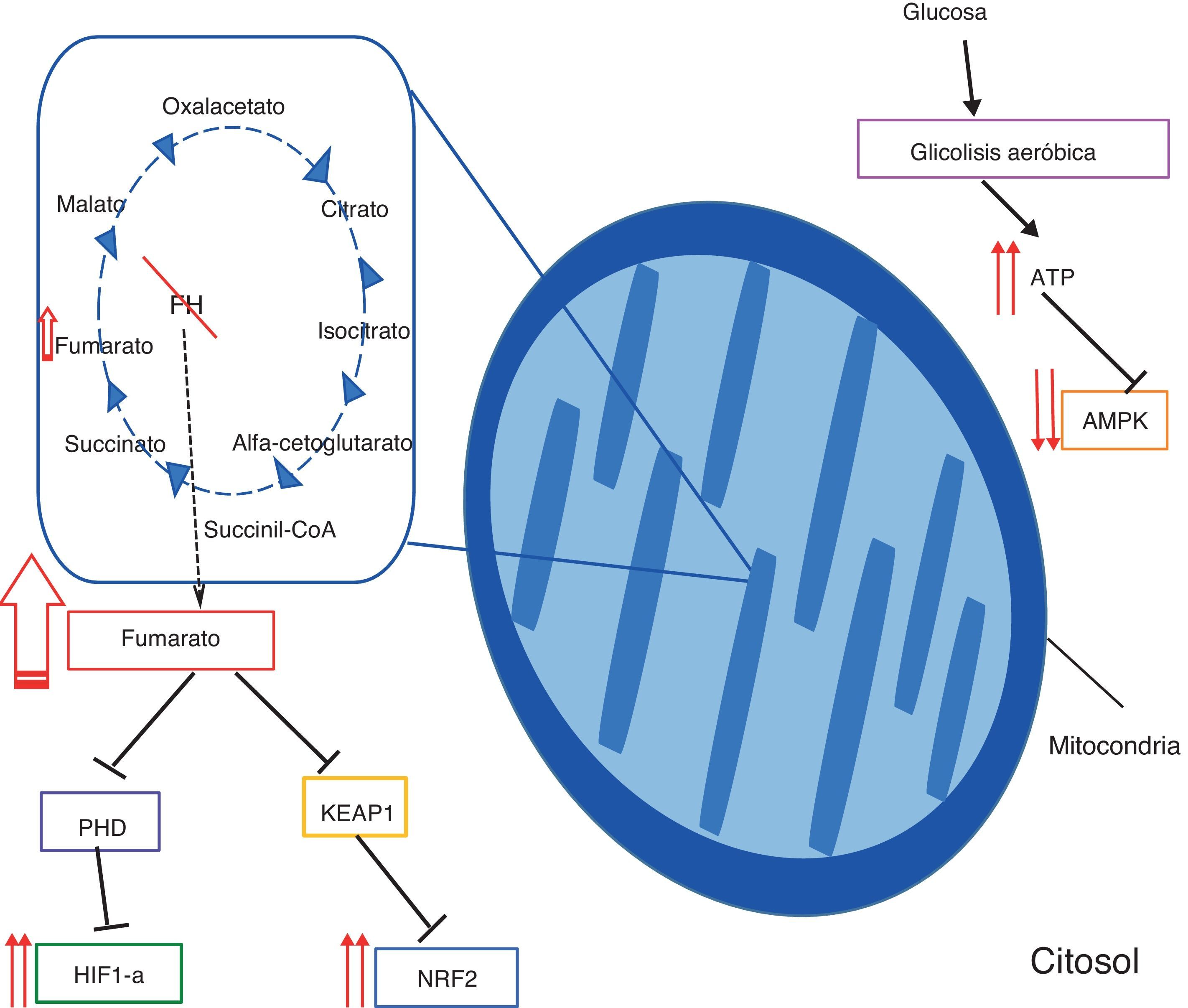
Esta proteína presenta 2 isotipos, uno citosólico del que se desconoce su función, pero podría estar relacionado en el metabolismo de aminoácidos; y uno mitocondrial que cataliza la conversión de fumarato en malato en el ciclo de Krebs2 (fig. 1).

Fisiopatología del HLRCC. Se resaltan en diferentes recuadros los principales mecanismos patológicos que se cree conllevan a la transformación maligna de las células deficientes en FH. La acumulación de fumarato entre otras, se relaciona con un cambio en la obtención de energía, que se realiza preferentemente vía glucólisis aeróbica, lo que conlleva un incremento del ATP y una disminución de AMPK. Por otro lado, las altas concentraciones de fumarato implican la inhibición de las proteínas PHD y KEAP1, lo que se traduce en una sobreactivación de las vías de HIF-a y NRF2 respectivamente.
Hasta la fecha, se han descrito más de 120 variantes patogénicas en el gen FH, siendo la mayoría de estas mutaciones de tipo missense; no obstante, pueden también encontrarse mutaciones nonsense, frameshift, insersiones/deleciones, del sitio de splicing y deleciones génicas de gran tamaño2,8,9.
En FH parecería existir además un hot spot mutacional en la localización Arg190, donde se han evidenciado múltiples mutaciones, siendo la c.698G>A la más común y apreciada especialmente en familias procedentes de Norteamérica e Inglaterra2,3,10.
Por otro lado, las siguientes mutaciones han sido observadas en mayor medida en familias afectadas por el cáncer renal: c.301C>T, c.302G>C, c.395T>C, c.698G>A, c.698G>T, c.697C>T, c.952C>T y c.233del. Sin embargo, el hecho de que los tumores renales no siempre se desarrollen en los portadores de estos cambios genéticos ilustra cuán complicado es delinear una relación genotipo-fenotipo clara en esta enfermedad2,3,10–12.
Fisiopatología del síndrome de leiomiomatosis y cáncer renal hereditarioEn los pacientes afectados por el síndrome se cree que hay una pérdida casi completa de la actividad de la fumarato hidratasa, lo cual se traduce en una acumulación de fumarato intracelular9,13,14 que a su vez genera 4 efectos patológicos principales (fig. 1).
- 1.
Hay una alteración del funcionamiento del ciclo de Krebs y produce un estado de seudohipoxia, lo que conduce a que se dé el efecto Warburg13,14, que implica que las células tumorales deficientes de FH suplen sus requerimientos energéticos mediante glucólisis aeróbica en lugar de utilizar fosforilación oxidativa8,15.
- 2.
Hay un aumento de las concentraciones del hypoxia-inducible factor 1- alpha (HIF1-a): Las altas concentraciones de fumarato en el citosol inhiben competitivamente a las enzimas prolyl-hydroxylases (PHD), las que normalmente hidroxilan los residuos de prolina del HIF1-a 9, con el fin de que un complejo con actividad ubiquitin ligasa, llamado von Hippel-Lindau protein (pVHL), marque al HIF1-a para degradación14.
Cuando las PHD se inhiben, el HIF1-a no se degrada apropiadamente y se da una sobreactivación de sus genes blancos que incluyen a platelet-derived growth factor (PDGF), glucose transporter 1 (GLUT1), vascular endothelial growth factor A (VEGF) y transforming growth factor alpha (TGF-a) y que se relacionan con procesos de proliferación celular, glucólisis en los tejidos y angiogénesis (fig. 1)9,14.
- 3.
Se produce una succinización aberrante con una mayor activación del nuclear factor erythroid 2-related factor 2 (NRF2): La succinización es una reacción favorecida por el exceso de fumarato13,16,17, que durante este proceso es capaz de interactuar con los grupos cisteína sulfhidrilo de las proteínas y por medio de una reacción de adición de Michael genera una modificación química estable llamada 2-succinyl-cysteine (2SC), que altera la estructura proteica e implica una inactivación funcional de la proteína blanco13.
La succinización mediada por fumarato tiene entonces como consecuencia directa la inhibición de la Kelch-like ECH-associated protein 1 (KEAP1)16, principal encargada de la degradación de NRF2 (2q31), que es un factor de transcripción perteneciente a la familia de cremalleras de leucina18 y actúa controlando la expresión de un grupo de genes con funciones citoprotectoras, antioxidantes y detoxificantes, los cuales tienen en su promotor un antioxidant response element (ARE) al que NRF2 se fija19–21. El gen que codifica este factor posee 5 exones y produce una proteína de 597 aminoácidos19,20.
En este sentido, la vía de NRF2 (fig. 2) es inducible y se encarga de proteger a las células frente al daño oxidativo, electrofílico y aquel producido por xenobióoticos20.
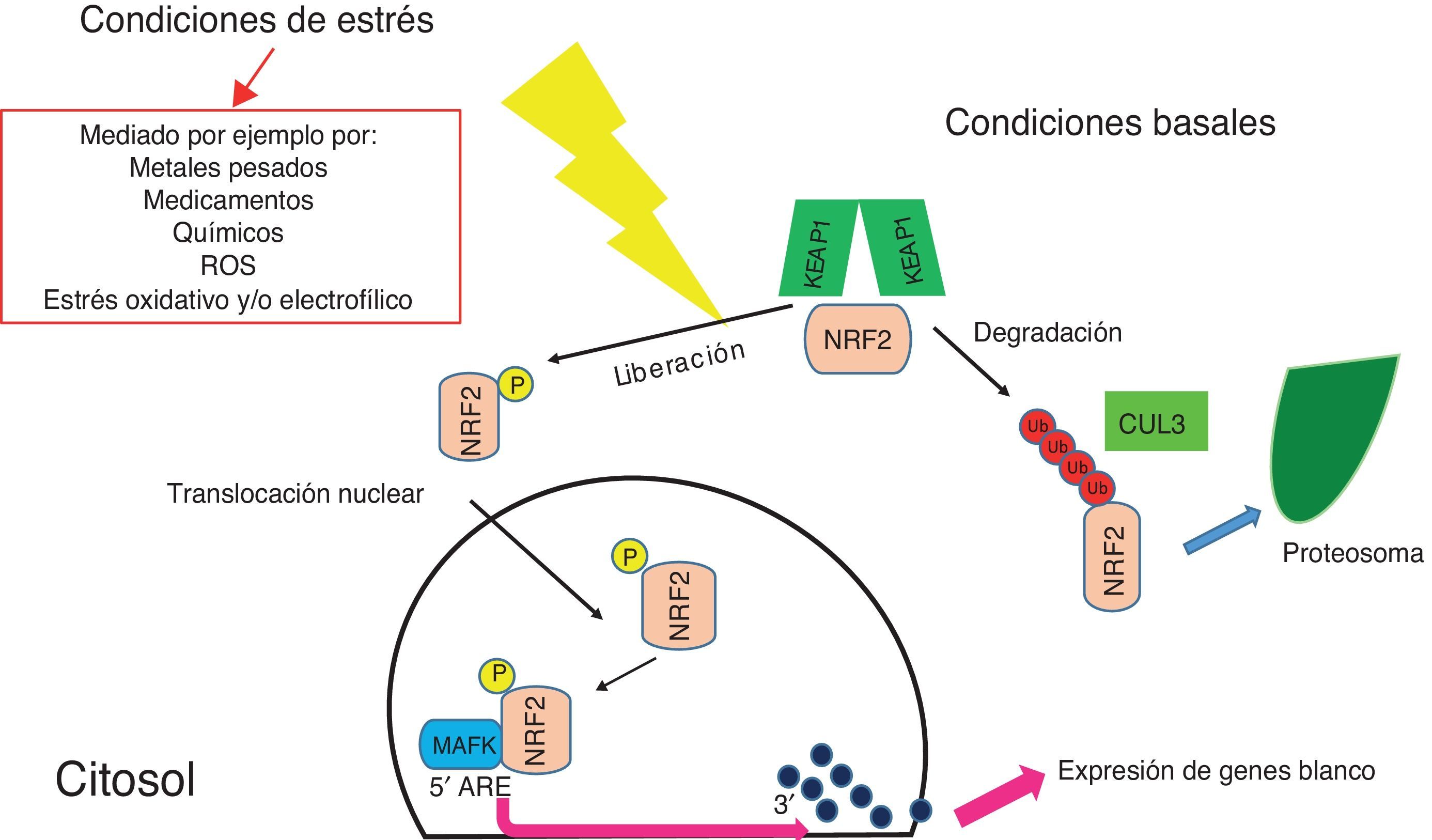
 Figura 2.
Figura 2.Visión básica de la vía de NRF2. En condiciones basales, NRF2 se encuentra acoplado a KEAP1 y constantemente es degradado por el proteosoma, gracias a la acción de CUL3. Cuando se produce algún tipo de estrés, mediado por diferentes fuentes como las enunciadas en el recuadro, NRF2 se libera de KEAP1, se fosforila y se transloca al núcleo donde se une al promotor de sus genes blancos, específicamente a un ARE; para ello requiere de la interacción con las proteínas MAFK.
(0.22MB).En condiciones normales, cuando no existe ningún tipo de estrés celular, NRF2 se encuentra en el citoplasma, sujeto a constante degradación, gracias a la estabilización que sufre cuando se une a KEAP1, proteína que funciona como un adaptador para que Cullin 3 (CUL3) –una ubiquitin ligasa 3– marque a NRF2 para degradación por el proteosoma (fig. 2)19,22.
En condiciones de estrés, que puede ser iniciado por diferentes daños en la célula (fig. 2), NRF2 se disocia de KEAP1 y escapa de la degradación19–22 para luego translocarse al núcleo, donde con ayuda de proteínas musculo-aponeurotic fibrosarcoma (MAFK), es capaz de unirse al elemento ARE del promotor sus genes blanco19–23.
La importancia de NRF2 en la fisiopatología del HLRCC estriba en que se le han atribuido acciones tanto antitumorales como protumorales. Las primeras tienen que ver con que es capaz de inducir vías de citoprotección celular, contra la toxicidad derivada de reactive oxygen species (ROS), estrés electrofílico, radiación, fármacos y toxinas15,21,22,24. De otra parte, la sobreexpresión de NRF2 y algunos de sus genes corriente abajo pueden favorecer procesos de malignización celular al aumentar rutas de supervivencia celular15,18,24–26, crecimiento celular27,28, metástasis24 y quimiorresistencia24,29,30,31.
Luego NRF2 tendría un rol dual en la célula. La actividad basal controlada de este factor de transcripción promovería la homeostasis celular y prevendría la malignización de los tejidos, ya que ayuda contrarrestar el daño en el ADN y a activar rutas de respuesta al estrés celular agudo. Sin embargo, la amplificación patológica de la señal de NRF2 –como ocurre en el contexto del HLRCC, debido a la succinización de KEAP1– facilitaría la transformación maligna de los tejidos al incrementar procesos protumorales20,30,32–34.
- 4.
Se alteran los niveles del sensor energético AMP-activated protein kinase (AMPK) los cuales se encuentran disminuidos, por razones aún no claras del todo15.

La compresión de la fisiopatología del HLRCC puede ayudar eventualmente al descubrimiento de mejores quimioterapéuticos35–37, que tengan como blancos terapéuticos a algunas de las proteínas ya mencionadas, siendo una de las más prometedoras NRF2 y algunos de sus genes corriente abajo.
Aspectos clínicosLeiomiomas cutáneosMás de un 75% de los afectados (portadores de la mutación en FH) pueden presentarlos a una edad promedio de 25 años. Son una serie de lesiones papulares o nodulares, de color café claro o eritematosas, que se distribuyen preferentemente en dorso, tronco y extremidades. Su diámetro aproximado es 0,4 y 2,5mm, aunque tienden a aumentar su número y tamaño con la edad2,4.
En la mayoría de los casos los leiomiomas cutáneos son asintomáticos; no obstante, en ocasiones se acompañan de dolor o parestesias en la zona donde se ubiquen2,4. Histológicamente, se reconocen como cúmulos de células musculares lisas, con núcleos ovales alargados4.
Leiomiomas uterinosSon masas fibroides, que cursan con dolor pélvico y sangrados menstruales irregulares. Aparecen en un 90% de las mujeres con el síndrome, entre los 18 y 52 años. Pueden ser causa de histerectomía temprana (antes de los 40 años) y se presentan como lesiones múltiples de un diámetro aproximado de 1-10cm3,38.
A nivel histológico, se observan células multinucleadas o con un núcleo único alargado. En menos del 1% de los casos, estos leiomiomas pueden malignizarse y convertirse en leiomiosarcomas uterinos, los cuales presentan una histología atípica4,39.
Cáncer renal papilar tipo 2También llamado carcinoma renal asociado al HLRCC, es todo tumor con características histológicas papilares tipo 2 que se encuentre en un paciente con mutación de línea germinal en FH y sin leiomiomatosis renal; esto de conformidad con la clasificación para tumores del sistema renal del año 2016, de la Organización Mundial para la Salud (OMS) y de la Sociedad Internacional de Patología Urológica (ISUP por sus siglas en inglés)40.
Se trata probablemente de la manifestación más devastadora del síndrome. Ocurre en un 10-16% de los afectados, con una edad de diagnóstico entre los 40-44 años y se estima que puede aparecer en un 20% de las familias41,42.
Son tumores unilaterales, dependientes de altas concentraciones de glucosa, con un alto potencial metastásico, por lo que la supervivencia promedio es de 5 años luego del diagnóstico41–43.
Su diagnóstico es casi siempre incidental y basado en el hallazgo de una masa renal en una resonancia magnética nuclear42,43. Empero, estos tumores pueden acompañarse de la triada dolor de espalda, hematuria macroscópica y masa palpable, la cual se registra en un 6-10% de los pacientes con algún tipo de cáncer renal44.
Respecto a la histología, los tumores se caracterizan por tener un citoplasma abundante, núcleos grandes y nucléolos prominentes que se encuentran rodeados por un halo claro2,4,40,42.
Otras manifestacionesSe han descrito además pacientes con: adenomas adrenales corticales, tumores de ducto colector renal, incidentalomas adrenales, cáncer de vejiga, seno, testículo, cistoadenomas de ovario42,45. Asimismo, es común la aparición de quistes renales4,46 que pueden ser el primer estadio de carcinogénesis en el tejido renal16.
DiagnósticoInicia por la sospecha clínica y requiere de una historia clínica y un examen físico minucioso2,4,43. Al igual que ocurre en otros trastornos de predisposición a cáncer, resulta relevante hacer énfasis en los antecedentes familiares de por lo menos 3 generaciones, anotando los miembros afectados por tumores renales u otros, leiomiomas cutáneos y/o uterinos; con la respectiva edad de aparición, evolución y síntomas asociados.
En el caso de pacientes afectados por neoplasias, debe además registrarse si esta fue la causa de muerte y a qué edad se produjo la misma.
Si bien se han propuesto una serie de criterios de diagnósticos para esta enfermedad, aún sigue siendo controversial su implementación y no existe un claro consenso sobre los mismos. En la tabla 1 presentamos un paralelo de los criterios propuestos por Smit et al. en el 20112 y luego por Schmidt et al.4, en el año 2014.
Comparación de los criterios diagnósticos propuestos para el síndrome HLRCC en los últimos años
| Criterios diagnósticos propuestos por Smit et al.2 | Criterios diagnósticos propuestos por Schmidt et al.4 |
|---|---|
| Criterio mayor | Criterio mayor |
| Múltiples leiomiomas cutáneos confirmados histopatológicamente | Múltiples leiomiomas cutáneos con al menos uno confirmado histopatológicamente |
| Criterios menores | Criterios menores |
| Tratamiento quirúrgico por leiomiomas uterinos sintomáticos antes de los 40 años | Leiomioma cutáneo solitario e historia familiar de HLRCC |
| Cáncer renal papilar tipo 2 antes de los 40 años | Múltiples leiomiomas uterinos sintomáticos de inicio temprano, antes de los 40 años |
| Un familiar en primer grado que tenga alguno de los anteriores criterios | Inicio temprano de tumores papilares tipo 2 antes de los 40 años |
| Diagnostico probable | Diagnostico definitivo |
| El probando tiene el criterio mayor | Análisis de mutación para FH positivo |
| Diagnostico de sospecha | |
| El probando tiene al menos 2 criterios menores | |
Llama la atención en estos criterios el que se tome como criterio mayor la presencia de leiomiomas cutáneos; que si bien son la manifestación más frecuente en los afectados, puede ser una de las más ambiguas, y es que de acuerdo con nuestra experiencia este tipo de lesiones cutáneas son difíciles de caracterizar, a menudo pueden pasar desapercibidas y en ocasiones simplemente estar ausentes5,11.
Aunado a lo anterior, el que los leiomiomas uterinos sean per se algo común en la población femenina general y que el cáncer renal papilar tipo 2 se evidencie en menos del 20% de los pacientes, contribuye a lo que Smit y otros2,47 denominan como el subdiagnóstico del HLRCC. Subdiagnóstico que solo puede contrarrestarse con un adecuado conocimiento del síndrome y sus particularidades, no solo por parte de los médicos especialistas, sino también por los médicos de atención primaria que son el contacto inicial de los pacientes con los servicios de salud y quienes pueden tomar la decisión de remitir a un individuo.
De igual forma, el papel de los patólogos es vital48,49, pues del reconocimiento de la morfología única que tienen los tumores renales papilares tipo 2 y los leiomiomas tanto cutáneos como uterinos depende en gran medida el diagnóstico definitivo de un paciente50. Con el fin de mejorar esta interpretación histopatológica y a partir de lo hallado por Bardella et al.13, se ha intentado con buenos resultados la inmunohistoquímica del 2SC como biomarcador de mutación en FH; la versatilidad de esta técnica permite que sea aplicable tanto a biopsias de tejido renal13,51, como cutáneo52,53 y uterino50,54; no obstante aún no está disponible en el ámbito clínico.
Pero, lejos de si se cumplen o no los criterios diagnósticos, o de si existe confirmación histológica, todo paciente con clínica sugestiva de HLRCC requiere remisión a genética, entre otras causas para iniciar los estudios moleculares confirmatorios y descartar otros diferenciales.
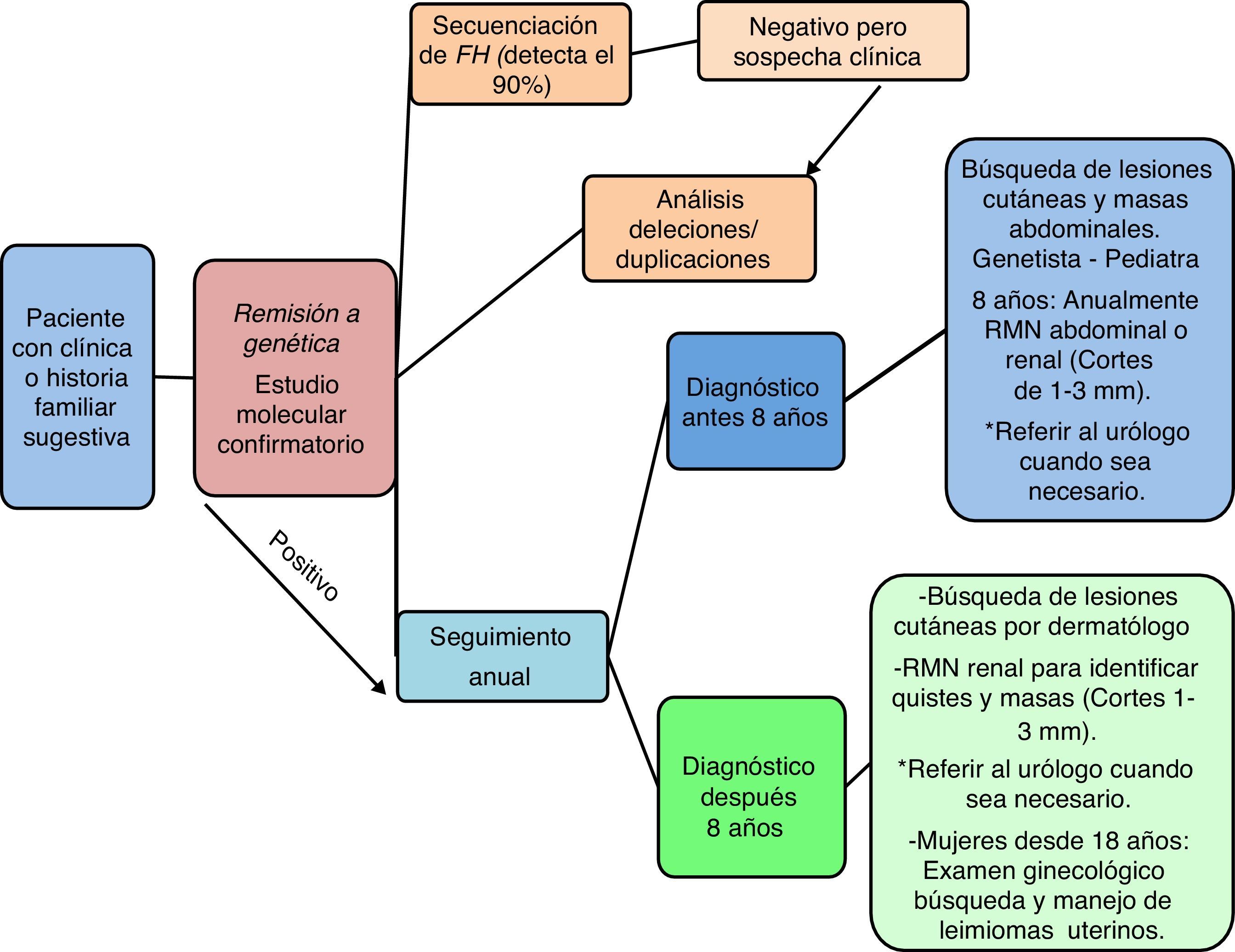
Diagnóstico genético y seguimientoSe debe iniciar siempre con una secuenciación completa del gen FH, abordaje que puede detectar más del 90% de los casos2,4. Ante un resultado negativo, pero una alta sospecha clínica, se optará por implementar un análisis de deleciones/duplicaciones, el cual puede realizarse con la ayuda de un multiplex ligation-dependent probe amplification (MLPA)55, una reacción en cadena de la polimerasa (PCR) cuantitativa o una hibridación genómica comparativa2,4,56. Todas estas técnicas se encuentran disponibles en nuestro país.
La confirmación molecular es absolutamente esencial para brindar una adecuada asesoría genética a las familias, y permite la implementación de un plan de seguimiento para los portadores de la mutación2,4,42 de acuerdo a la edad de diagnóstico (fig. 3); que puede ser inclusive antes de los 8 años de edad57 como lo estipula la organización HLRCC Alliance58. De manera que, en menores de 8 años de edad se debe realizar una concienzuda búsqueda de masas abdominales con el fin de identificar tumores renales u otros, por parte del genetista y/o pediatra; la resonancia magnética nuclear renal se programará anualmente 42,58,59. En mayores de 8 años, además de una activa vigilancia frente a tumores renales, se debe descartar la presencia de leiomiomas cutáneos y, en mujeres, la de leiomiomas uterinos a partir de los 18 años de edad, momento en el que deben referirse a ginecología58. En cualquier caso, la resonancia magnética nuclear renal debe hacerse teniendo en cuenta que los cortes usados durante el procedimiento deben oscilar entre 1 y 3mm58, esto debido a que las masas renales aún cuando son muy pequeñas pueden asociarse a metástasis, por lo que es absolutamente relevante su identificación temprana. Ante el hallazgo de un tumor renal, la interconsulta con urología es prioritaria, con el fin de iniciar lo más rápido posible el manejo y se recomienda la realización de una tomografía computarizada abdominal complementaria para delinear los márgenes del tumor42.
Tratamiento
El manejo debe ser por parte de un equipo multidisciplinario60, que incluya genetista, dermatólogo, ginecólogo, urólogo y oncólogo.
En el caso de los leiomiomas cutáneos, generalmente se da un tratamiento sintomático del dolor y las parestesias asociadas, con medicamentos como gabapentina, nifedipino y nitroglicerina4. La remoción quirúrgica está indicada para lesiones únicas, y ante formas múltiples se prefieren las terapias con crioterapia, electrocoagulación o ablación laser42,61,62. Habrá que tener en cuenta que en un 50% de los casos, estas lesiones pueden recurrir60.
Respecto a los leiomiomas uterinos, es el ginecólogo el que de acuerdo a la gravedad de la sintomatología evalúa la necesidad de iniciar anticonceptivos orales para mejorar el sangrado y el dolor pélvico, de realizar un abordaje conservador con miomectomía o de efectuar una histerectomía en casos graves2–4,8.
En cuanto a los tumores renales papilares tipo 2, dada su agresividad inclusive cuando son menores a 1cm, requieren siempre de tratamiento quirúrgico, que puede ser nefrectomía parcial o total, más linfadenectomía retroperitoneal42 seguida de quimioterapia con bevacizumab y erlotinib4.
DiscusiónEl síndrome HLRCC es una enfermedad rara, que cursa con leiomiomas cutáneos y uterinos, así como con tumores renales papilares tipo 2, estos últimos muy agresivos y de pobre pronóstico, que pueden aparecer inclusive antes de los 8 años de edad42,57.
Se trata de una enfermedad que se genera por mutaciones de línea germinal en el gen FH, que produce la proteína fumarato hidratasa, la cual posee un rol esencial en el ciclo de Krebs, durante la conversión de fumarato en malato14. Actualmente la hipótesis más aceptada, respecto al porqué se genera la transformación maligna de los tejidos en los afectados, gira en torno a la noción de que es la concentración excesiva de fumarato en la célula la responsable de la sobreactivación de los genes corriente abajo de los factores de transcripción HIF-1a y NRF215; siendo este último uno de los investigados hoy en día por el potencial rol que tendrían algunos de los genes de su vía, como biomarcadores63 y blancos terapéuticos64,65 en los afectados por el cáncer renal, donde se hace necesario encontrar nuevas formas de tratamiento que logren mejorar la supervivencia.
Respecto al diagnóstico, este es de sospecha y se precisa siempre una confirmación molecular adecuada66, por lo que es primordial la evaluación por un genetista.
Asimismo, es necesario un equipo multidisciplinario que trabaje mancomunadamente, para realizar un adecuado seguimiento y tratamiento, que en el caso de los leiomiomas cutáneos y uterinos es principalmente sintomático4.
Mención especial merece el tratamiento de los tumores renales, ya que a diferencia de otros síndromes de predisposición a cáncer, en el HLRCC no se aplica la «regla de los 3cm» para realizar una resección tumoral, ya que las neoplasias exhibidas por los pacientes tienen un crecimiento muy rápido con metástasis asociada, por lo que ante cualquier masa renal, el manejo es quirúrgico y no se recomienda una conducta expectante42,67. Por otro lado, dado que se trata de tumores unilaterales, la nefrectomía radical puede estar indicada cuando se sospecha que aún con márgenes amplios de resección y linfadenectomía retroperitoneal una nefrectomía parcial no será curativa42. Y es que, en términos generales, los tumores renales asociados al HLRCC tienen un comportamiento muchísimo más agresivo que los relacionados con enfermedad de von Hippel Lindau o síndrome Birt–Hogg–Dubè42.
Finalmente, resulta importante mencionar que si bien el HLRCC es una enfermedad muy poco frecuente, de la que inclusive no se cuenta con datos de incidencia o prevalencia47 y que no ha sido objeto de estudio en Colombia u otros países de Latinoamérica –donde creemos que la primera familia reportada es aquella descrita por nuestro grupo en el año 20165– es ilógico pensar que no existan otros individuos afectados en el país. Máxime, si tenemos en cuenta el subdiagnóstico de este trastorno, ampliamente comentado por algunos autores2,47 y que puede deberse al desconocimiento del síndrome como tal, por parte de los médicos de atención primaria y especialistas, que no reconocen los leiomiomas cutáneos y no asocian los leiomiomas uterinos de inicio temprano con un trastorno hereditario. De igual manera, el hecho de que los tumores renales papilares tipo 2 aparezcan solo en el 10-16% de los afectados, y que a veces sean difíciles de caracterizar histológicamente, solo empeora el escenario. Así que, conocer las peculiaridades clínicas y moleculares de este síndrome es fundamental para el reconocimiento y manejo de los afectados, quienes sufren enormemente por la falta de un diagnóstico y tratamiento certeros.
ConclusionesEl síndrome HLRCC es una enfermedad caracterizada por la presencia de leiomiomas cutáneos, uterinos y cáncer renal papilar tipo 2. Los pacientes y sus familias se benefician de una intervención temprana, una asesoría genética integral y del manejo por parte de un equipo multidisciplinario. Es un trastorno bastante raro, pero en el que no puede descartarse un subdiagnóstico, que solo puede vencerse aumentando el conocimiento de este síndrome en los médicos generales y especialistas.
Conflicto de interesesLas autoras declaran no tener ningún conflicto de intereses.
AgradecimientosAl doctor Carlos Arturo Florido Caicedo, director del Departamento de Morfología de la Universidad Nacional de Colombia, por toda su ayuda en la consecución de este trabajo y por su especial colaboración en la creación de las figuras presentadas.