




Despite the availability of several COVID-19 vaccines, the incidence of infections remains a serious issue. Tunicamycin (TM), an antibiotic, inhibited tumor growth, reduced coronavirus envelope glycoprotein subunit 2 synthesis, and decreased N-linked glycosylation of coronavirus glycoproteins.
ObjectivesOur study aimed to determine how tunicamycin interacts with certain coronavirus proteins (proteinase, protease, nsp9, ORF7a, ORF3a, ORF9b, ORF8, envelope protein, nsp2, and RBD of spike glycoprotein). Methods: Several types of chemo and bioinformatics tools were used to achieve the aim of the study. As a result, virion's effectiveness may be impaired.
ResultsTM can bind to viral proteins with various degrees of affinity. The proteinase had the highest binding affinity with TM. Proteins (ORF9b, ORF8, nsp9, and RBD) were affected by unfavorable donor or acceptor bonds that impact the degree of docking. ORF7a had the weakest affinities.
ConclusionsThis antibiotic is likely to effect on SARS-CoV-2 in clinical studies.
A pesar de la disponibilidad de varias vacunas contra la COVID-19, la incidencia de infecciones sigue siendo un problema grave. La tunicamicina (TM), un antibiótico, inhibió el crecimiento tumoral, redujo la síntesis de la subunidad 2 de la glicoproteína de la envoltura del coronavirus y disminuyó la glicosilación ligada a N de las glicoproteínas del coronavirus.
ObjetivosNuestro estudio tuvo como objetivo determinar cómo interactúa la tunicamicina con ciertas proteínas del coronavirus (proteinasa, proteasa, nsp9, ORF7a, ORF3a, ORF9b, ORF8, proteína de la envoltura, nsp2 y RBD de glicoproteína de punta).
MétodosSe utilizaron varios tipos de herramientas de quimioterapia y bioinformática para lograr el objetivo del estudio. Como resultado, la eficacia del virión puede verse afectada.
ResultadosLa TM puede unirse a proteínas virales con diversos grados de afinidad. La proteinasa tenía la mayor afinidad de unión con TM. Las proteínas (ORF9b, ORF8, nsp9 y RBD) se vieron afectadas por enlaces donantes o aceptores desfavorables que afectan el grado de acoplamiento. ORF7a tenía las afinidades más débiles.
ConclusionesEs probable que este antibiótico tenga efecto sobre el SARS-CoV-2 en estudios clínicos.
SARS-CoV-2 is the most lethal of the coronaviruses, causing over 4.4 million deaths from coronavirus sickness 2019 (COVID-19) and affecting over 210 million people worldwide through the middle of August 2021. SARS-CoV-2 has a genome that is almost 30 kb long and has 14 open reading frames (ORFs) that encode 27 proteins. It has four structural proteins (surface, envelope, membrane, and nucleocapsid) as well as sixteen non-structural proteins (nsp1-16).1,2 Several antiviral medications are now being studied in clinical trials, but reputable clinical trials are becoming increasingly difficult to run as the public's need for easily available remedies grows. Remdesivir is an antiviral that is now being tested in clinical trials for the treatment of COVID-19. It works by inhibiting RNA synthesis by targeting RdRP.3–5 Paxlovid is the first oral antiviral medicine approved by the FDA for adults and children with mild to moderate COVID-19. Paxlovid is a ritonavir-boosted nirmatrelvir medication for those aged 12 and above who are at high risk of contracting severe COVID-19, which can lead to hospitalisation or death.6–8
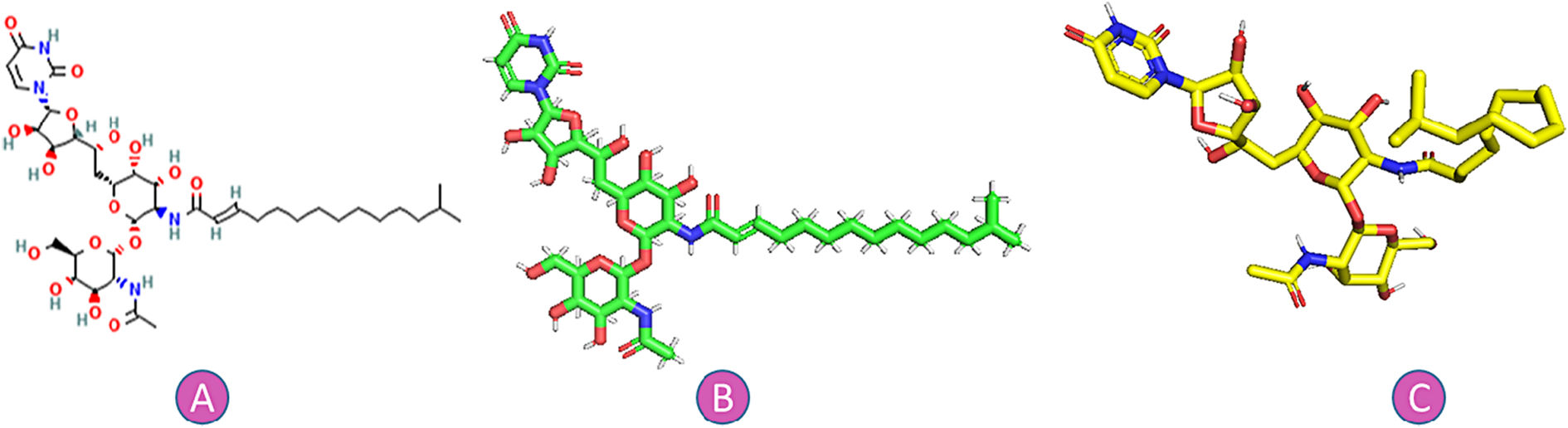
Tunicamycin (C38H62N4O16) is an antibiotic made by S. clavuligerus, S. lysosuperficus, and among other bacteria, Fig. 1. TM is a white crystalline powder that is mildly soluble in ethanol, acidic water, chloroform, and benzene, and is soluble in alkaline water and pyridine.9–11 There is currently no information available on TM's pharmacodynamics, absorption, toxicity, or metabolism.12–14

Tunicamycin (TM) causes endoplasmic reticulum stress in cells by blocking the initial step in the production of N-linked glycans in proteins, resulting in a high number of missfolded proteins.15,16 The cell cycle is stopped in the G1 phase when the antibiotic prevents glycosylation of N-glycans. Previous research showed that TM might be utilized to treat human colon and prostate cancer cells by inducing apoptosis.17,18
TM treatment significantly decreased tumor growth and significantly prolonged the lifetime of tumor-bearing mice in vitro compared to the PBS-treated group. TM has been shown to suppress the production of coronavirus envelope glycoprotein subunit 2 but not the synthesis or glycosylation of envelope glycoprotein subunit 1 during virion generation or release. Infected cells with TGEV in the presence of TM had significantly reduced antigenicity of both surface and membrane proteins.19,20 This might support the hypothesis that TM is a glycosylation protein inhibitor. Coronavirus generated spikeless, non-infectious virions that lacked S protein when cultured in the presence of the inhibitor TM. Furthermore, No evidence that TM inhibits N-linked glycans in nonstructural proteins (nsp3, and nsp4) has been found too far.21,22
In vitro, TM has been used to determine the functional importance of N-glycosylation in biological systems, such as cell proliferation and survival, drug sensitivity and resistance of tumor cells to anti-cancer medicines, and programmed cell death (apoptosis).23,24
TM induces DR5 expression in prostate cancer cells, making it a potent activator of TRAIL (Tumor necrosis factor–related apoptosis–inducing ligand)-induced apoptosis. Furthermore, p53 is inactive in PC-3 and DU145 cells. As a result, p53-deficient tumor cells may benefit from a combination of TM and TRAIL treatment. It's critical to know if TM and TRAIL can be given safely without producing toxicity in normal tissues such as the liver. This data implies that tumor vs normal cell susceptibility to the TM sensitization effect is connected to CHOP's (C/EBP homologous protein) uneven induction capability. These findings suggest that combining TM with TRAIL to treat hormone-refractory prostate cancer might be beneficial.25 On the surface of tumor cells, TM reduced the amount of lectin-binding sites. Not only in parental cells, but also in generated cisplatin-resistant cells, TM enhances in vitro cisplatin sensitivity. In clinical studies, TM has shown to be effective in overcoming cisplatin resistance.26 Moreover, TM prevents the production of the MHV E2 glycoprotein but not the synthesis or glycosylation of the transmembrane glycoprotein El, viriogenesis, or viriolysis from cells.27
The goal of our study was to fig. out how tunicamycin interacts with specific coronavirus proteins. On the other hand, finding the docking degree of the TM with the protein may be important in determining the likelihood of employing this antibiotic to suppress the virus in clinical trials on mice.
MethodsThe reference sequence SARS-CoV-2 (NC 045512.2) was selected as the source of all proteins utilized in this study as follows: Proteinase 3CLpro (PDB: 1P9S), protease (PDB: 1Q2W), nonstructural protein nsp9 (PDB: 1QZ8), ORF7a accessory protein (PDB: 1XAK), ORF3a (PDB: 6XDC), ORF9b (PDB: 7DHG), ORF8 encoded accessory protein (PDB: 7JX6), envelope protein (pentameric transmembrane domain PDB: 7K3G), nonstructural protein nsp2 (PDB:7MSW), and receptor-binding domain (RBD) of spike glycoprotein (PDB: 6M0J). The structure of tunicamycin was extracted from PubChem.
Prepare protein for dockingAfter downloading proteins from the Protein Data Bank and loading them into the BIOVIA Discovery Studio Visualizer program, each protein was passed. All water molecules and hetatoms were removed from the total protein after the hierarchical scale was created. To aid molecular fusion, hydrogen polar bonds were introduced.
Molecular docking between Tunicamycin and each viral proteinTo determine the interaction of tunicamycin with each protein, we used PyRx software for autodock. The protein structure was converted to macromolecule (pdbqt) type after loading the protein. TM was minimized energy and changed to pdbqt style after loading the legend. VINA wizard was used to conducting molecular docking. After minimizing energy, the entire interaction was carried out. To analyze the structural vision, the top three interactions were chosen.
Determine the molecular docking's 3D structureThe degree of contact between the protein and the ligand was calculated, and the interaction areas between atoms were carefully mapped. PyMOL software was used to extract the docking's 3D structure.
ResultsAccording to the findings of this study, tunicamycin can bind to SARS-CoV-2 proteins in a specific region depending on the kind and location of attachment. The PyRx picked 9 fusion models based on the degree of affinity and the quantity of energy based on the docking models for each protein. The top three docking models were chosen based on the highest protein-ligand affinity. Table 1 displays the top three models that can be retrieved after each protein binds tunicamycin. The protein with the highest binding affinity was chosen when (RMSD/ub) and (RMSD/lb) both equaled zero. RMSD/ub denotes the upper bound of the root mean square deviation, whereas RMSD/lb denotes the lower bound of the root mean square deviation.
Binding affinity and RMSD degree of the top of molecular docking models between viral proteins and TM. The highest binding affinity docking for each protein was selected (blue models).
| Protein-Ligand Docking | Binding Affinity | RMSD/ub | RMSD/lb |
|---|---|---|---|
| 1P9S_11104835_uff_E=1878.14 | -9.4 | 0 | 0 |
| 1P9S_11104835_uff_E=1878.14 | -9.3 | 8.812 | 4.721 |
| 1P9S_11104835_uff_E=1878.14 | -9.2 | 9.162 | 5.207 |
| 1Q2W_11104835_uff_E=1774.64 | -8.2 | 0 | 0 |
| 1Q2W_11104835_uff_E=1774.64 | -7.9 | 10.756 | 3.817 |
| 1Q2W_11104835_uff_E=1774.64 | -7.9 | 13.31 | 7.184 |
| 1QZ8_11104835_uff_E=1774.64 | -6.4 | 0 | 0 |
| 1QZ8_11104835_uff_E=1774.64 | -6.2 | 25.588 | 20.811 |
| 1QZ8_11104835_uff_E=1774.64 | -6.2 | 22.963 | 19.059 |
| 1XAK_11104835_uff_E=1774.64 | -4.9 | 0 | 0 |
| 1XAK_11104835_uff_E=1774.64 | -4.9 | 11.051 | 6.634 |
| 1XAK_11104835_uff_E=1774.64 | -4.9 | 13.824 | 5.495 |
| 6XDC_11104835_uff_E=1774.64 | -8.8 | 0 | 0 |
| 6XDC_11104835_uff_E=1774.64 | -8.5 | 6.972 | 4.478 |
| 6XDC_11104835_uff_E=1774.64 | -8.5 | 6.522 | 3.488 |
| 7DHG_11104835_uff_E=1774.64 | -7.8 | 0 | 0 |
| 7DHG_11104835_uff_E=1774.64 | -7.4 | 31.828 | 26.717 |
| 7DHG_11104835_uff_E=1774.64 | -7.4 | 9.123 | 4.383 |
| 7JX6_11104835_uff_E=1774.64 | -7 | 0 | 0 |
| 7JX6_11104835_uff_E=1774.64 | -7 | 9.401 | 3.118 |
| 7JX6_11104835_uff_E=1774.64 | -6.9 | 11.72 | 4.807 |
| 7K3G_model1_11104835_uff_E=1774.64 | -8 | 0 | 0 |
| 7K3G_model1_11104835_uff_E=1774.64 | -7.9 | 4.09 | 2.628 |
| 7K3G_model1_11104835_uff_E=1774.64 | -7.7 | 2.416 | 1.75 |
| 7MSW_11104835_uff_E=1774.64 | -8.1 | 0 | 0 |
| 7MSW_11104835_uff_E=1774.64 | -7.8 | 28.359 | 24.597 |
| 7MSW_11104835_uff_E=1774.64 | -7.7 | 2.452 | 2.009 |
| RBD_11104835_uff_E=1774.64 | -6.9 | 0 | 0 |
| RBD_11104835_uff_E=1774.64 | -6.8 | 2.528 | 1.817 |
| RBD_11104835_uff_E=1774.64 | -6.8 | 2.109 | 1.058 |
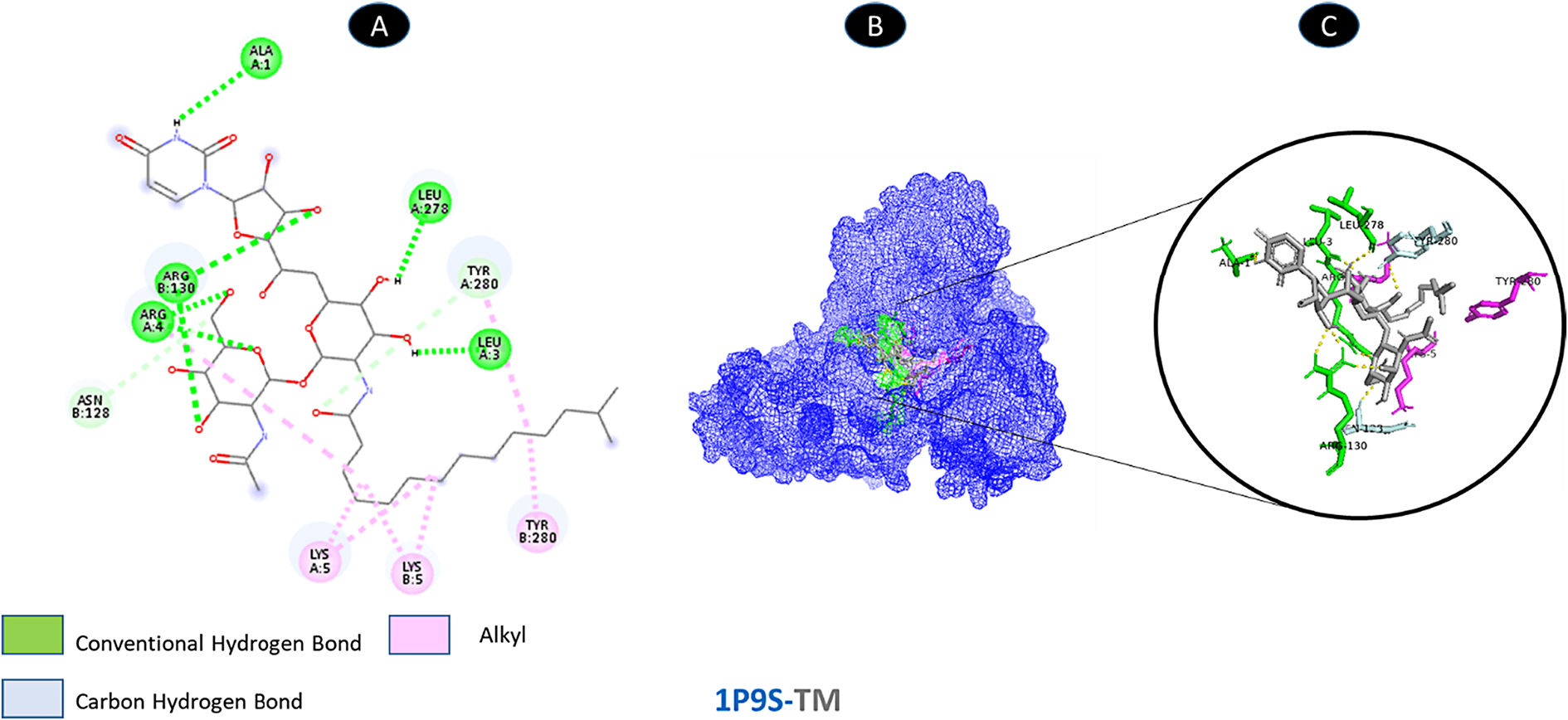
Tunicamycin and proteinase share 5 conventional hydrogen bonding residues (A: ALA-1, A: LEU-3, A: ARG-4, A: LEU-278, and B: ARG-130) and 2 carbon-hydrogen bonding residues (A: TYR-280, and B: ASN-28). There are three possible weakest alkyl interactions (A: LYS-5, B: LYS-5, and B: TYR-280) as illustrated in Fig. 2.
 shows the interaction location with TM. C: Molecular docking residues of 1P9S-TM, residues of conventional hydrogen bond (green), residues of carbon-hydrogen bond (cyan), and alkyl residues (magenta).")
Molecular docking of 1P9S-TM. A: 2D interaction shows types of fusion in specific residues. B: Crystal structure of proteinase (1P9S) shows the interaction location with TM. C: Molecular docking residues of 1P9S-TM, residues of conventional hydrogen bond (green), residues of carbon-hydrogen bond (cyan), and alkyl residues (magenta).
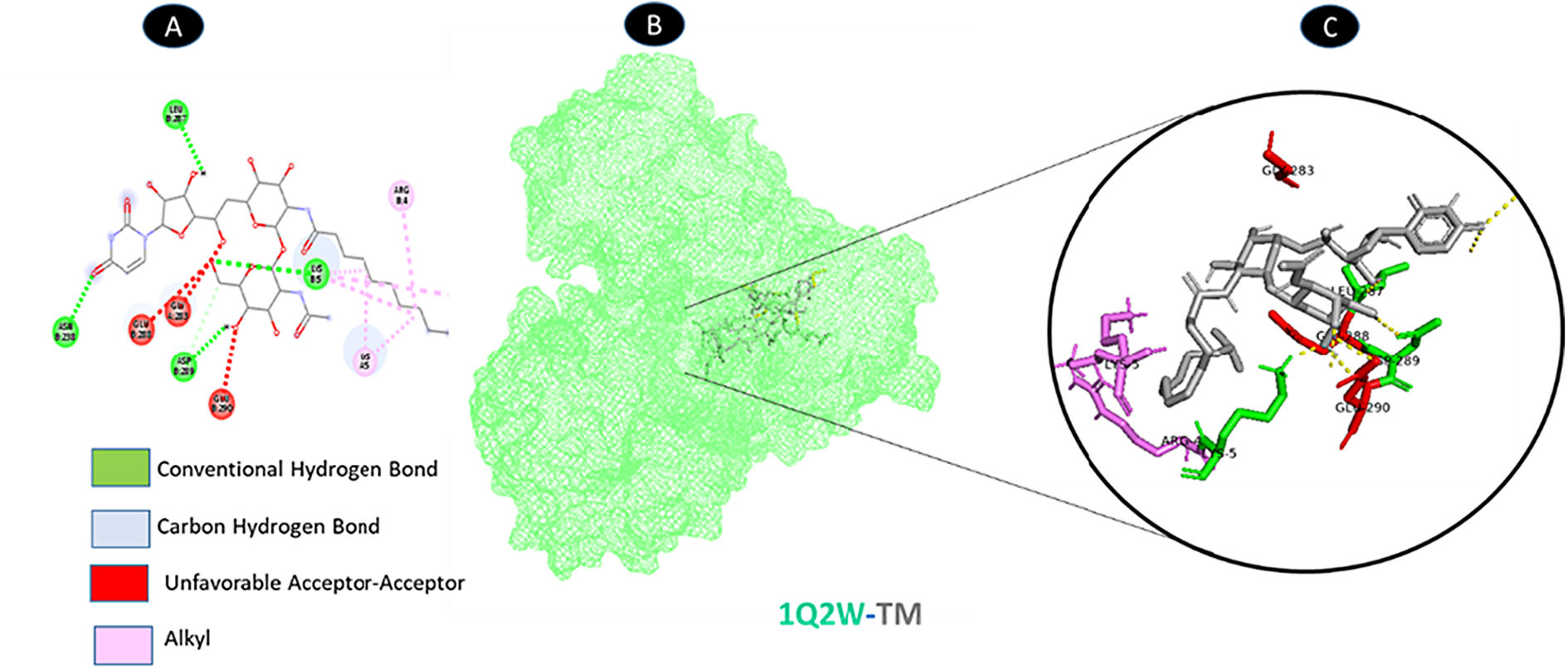
TM and protease share four typical hydrogen bonding residues (B: LYS-5, B: ASN-238, B: LEU-287, and B: ASP-289), as well as two of the weakest alkyl interactions (A: LYS-5, and B: ARG-4). As shown in Fig. 3, there are three unfavorable acceptor residues (A: GLY-283, B: GLU-288, and B: GLU-290).
 shows the interaction location with TM. C: Molecular docking residues of 1Q2W-TM, residues of conventional hydrogen bond (green), residues of carbon-hydrogen bond (cyan), alkyl residues (magenta), and unfavorable acceptor bound (red).")
Molecular docking of 1Q2W-TM. A: 2D interaction shows types of fusion in specific residues. B: Crystal structure of protease (1Q2W) shows the interaction location with TM. C: Molecular docking residues of 1Q2W-TM, residues of conventional hydrogen bond (green), residues of carbon-hydrogen bond (cyan), alkyl residues (magenta), and unfavorable acceptor bound (red).
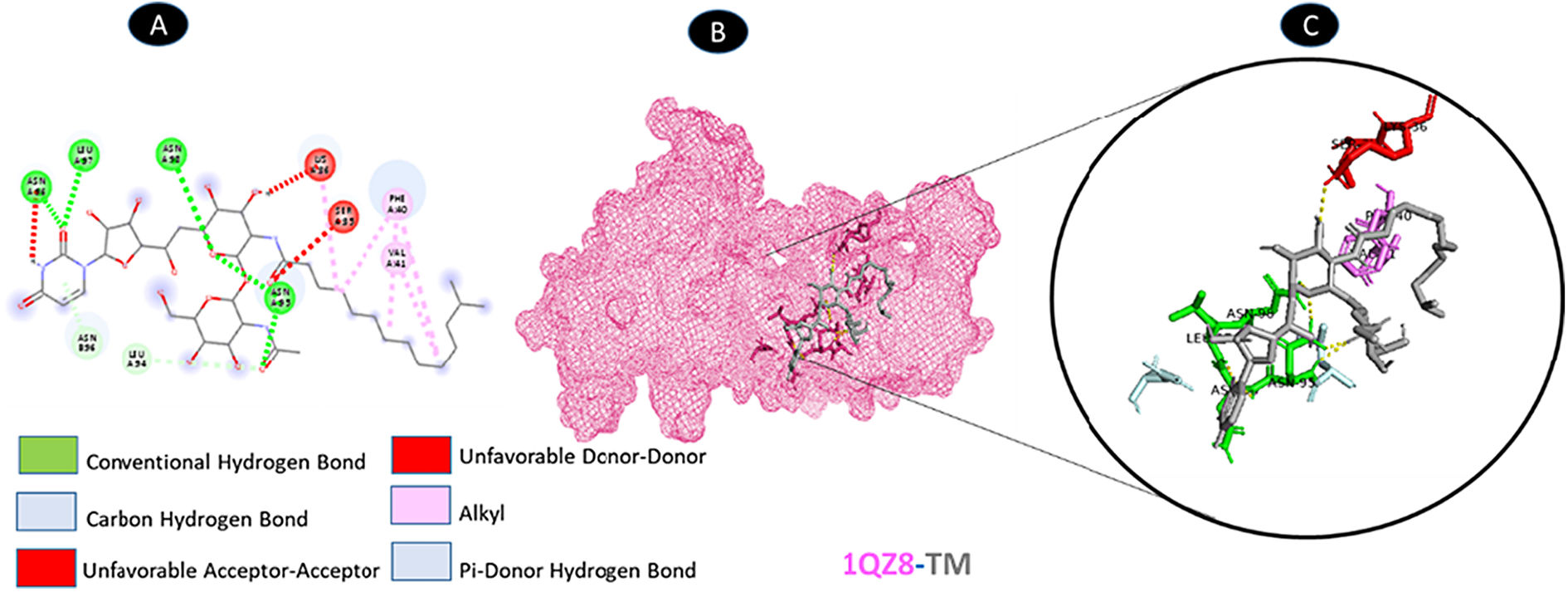
TM and nsp9 share four hydrogen bonding residues (A: ASN-95, ASN-96, A: LEU-97, and A: ASN-98) as well as two carbon-hydrogen bonding residues (A: LEU-94, and B: ASN-96). As shown in Fig. 4, there are two probable weakest alkyl interactions (A: PHE-40 and A: VAL-41) as well as two unfavorable acceptor or donor contacts (A: SER-95 and A: LYS-96).
 shows the interaction location with TM. C: Molecular docking residues of 1QZ8-TM, residues of conventional hydrogen bond (green), residues of carbon-hydrogen bond (cyan), alkyl residues (magenta), pi-donor hydrogen bond (dark cyan), and unfavorable acceptor or donor bond (red).")
Molecular docking of 1QZ8-TM. A: 2D interaction shows types of fusion in specific residues. B: The crystal structure of nsp9 (1QZ8) shows the interaction location with TM. C: Molecular docking residues of 1QZ8-TM, residues of conventional hydrogen bond (green), residues of carbon-hydrogen bond (cyan), alkyl residues (magenta), pi-donor hydrogen bond (dark cyan), and unfavorable acceptor or donor bond (red).
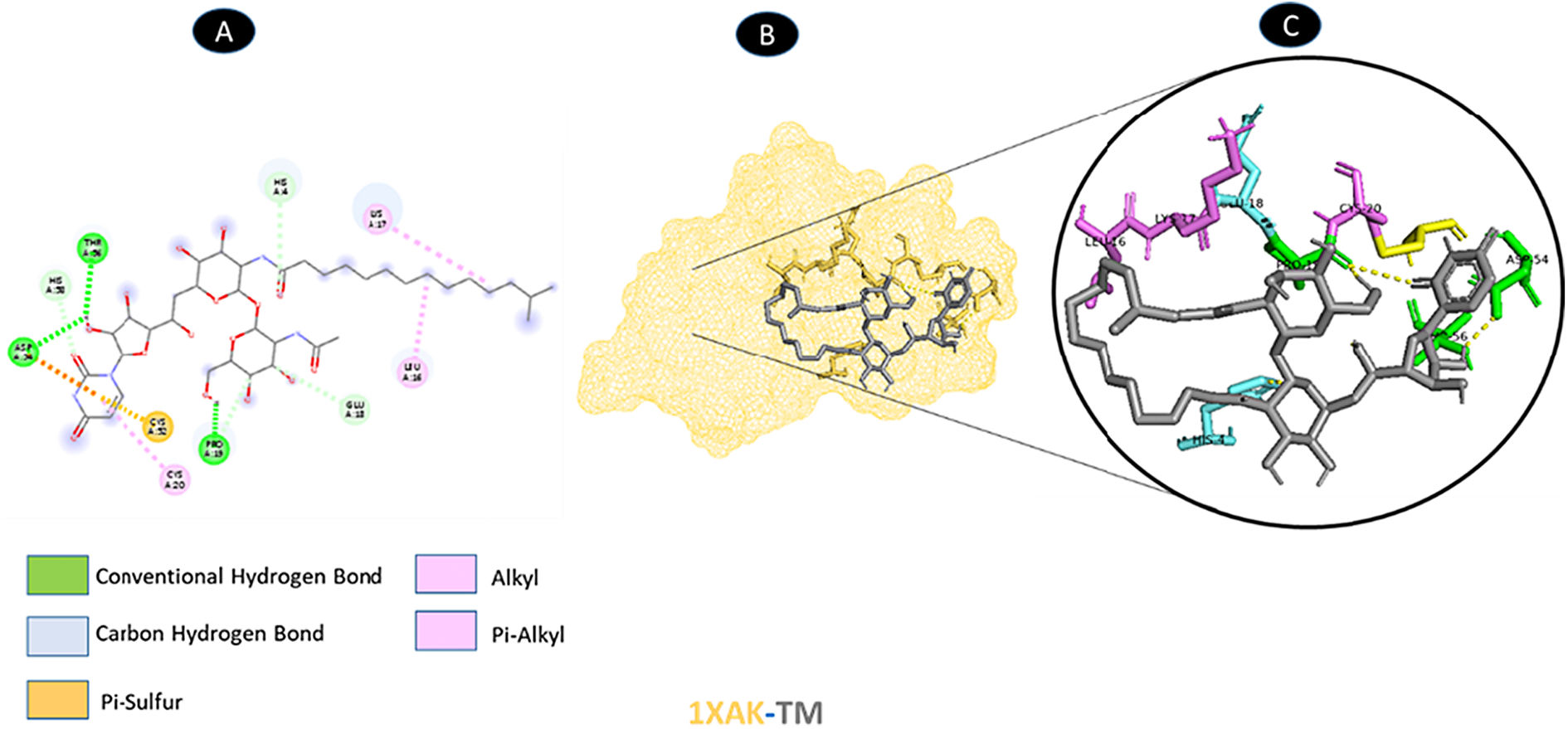
Three conventional hydrogen bonding residues (A: PRO-19, A: ASP-54, and A: THR-56), as well as 3 carbon-hydrogen bonding residues (A: HIS-4, A: GLU-13, and A: HIS-50), are shared by TM and ORF7a. As illustrated in Fig. 5, there are two probable weakest alkyl interactions (A: LEU-16, A: LYS-17, and A: CYS-20) as well as a pi-sulfur interaction residue (A: CYS-52).
 shows the interaction location with TM. C: Molecular docking residues of 1XAK-TM, residues of conventional hydrogen bond (green), residues of carbon-hydrogen bond (cyan), alkyl, and pi-alkyl residues (magenta), and pi-sulfur bond (pale orange).")
Molecular docking of 1XAK-TM. A: 2D interaction shows types of fusion in specific residues. B: The crystal structure of ORF7a (1XAK) shows the interaction location with TM. C: Molecular docking residues of 1XAK-TM, residues of conventional hydrogen bond (green), residues of carbon-hydrogen bond (cyan), alkyl, and pi-alkyl residues (magenta), and pi-sulfur bond (pale orange).
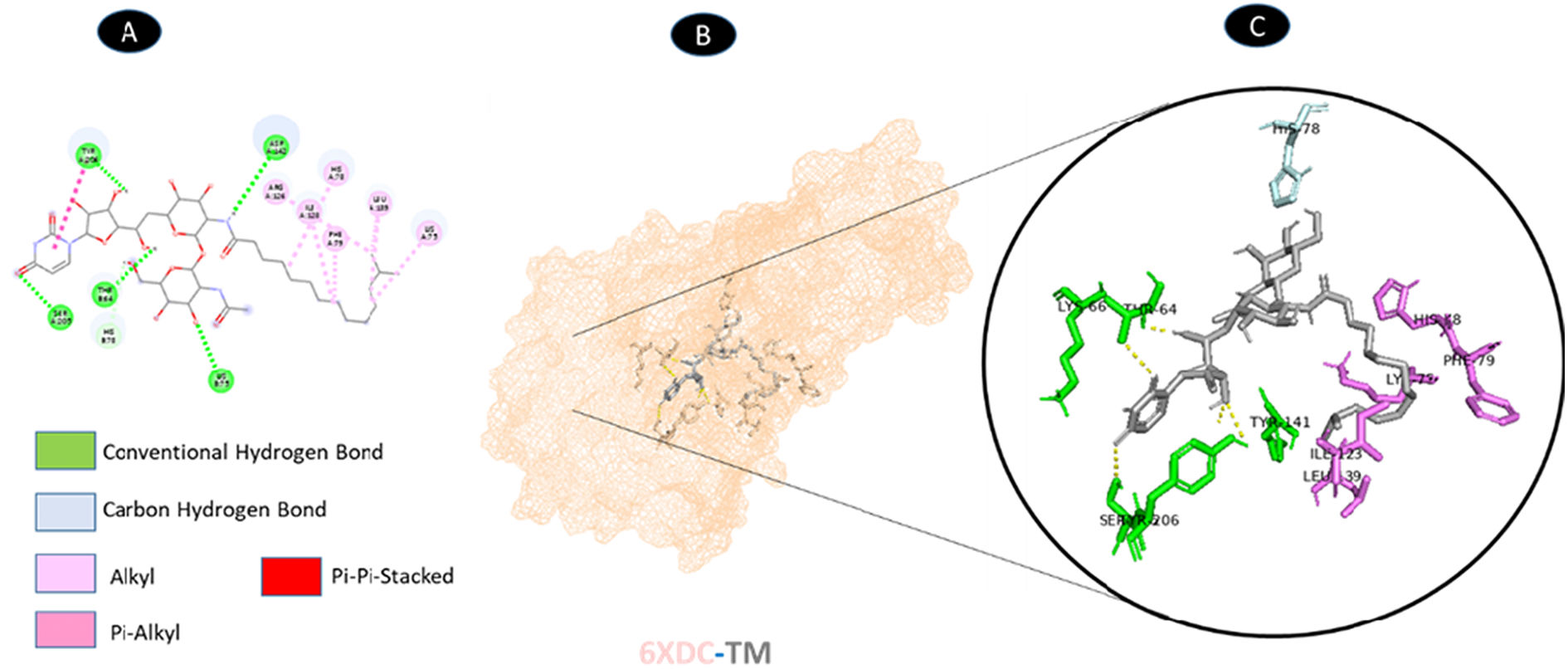
TM and ORF3a share 5 conventional hydrogen bonding residues (A: ASP-142, A: SER-205, A: TYR-206, B: THR-64, and B: LYS-75) as well as a carbon-hydrogen bonding residue (B: HIS-78). There are six possible weakest alkyl interactions (A: LYS-75, A: HIS-78, A: PHE-79, A: ARG-126, A: ILE-128, and A: LEU-139). As illustrated in Fig. 6, there is additionally one pi-alkyl bound in the residue (A: TYR-206) and 20 pi-pi-stacked bounds in a single or double.
 shows the interaction location with TM. C: Molecular docking residues of 6XDC-TM, residues of conventional hydrogen bond (green), residues of carbon-hydrogen bond (cyan), alkyl residues (magenta), pi-alkyl bond (dark magenta), and pi-pi-stacked bound (red).")
Molecular docking of 6XDC-TM. A: 2D interaction shows types of fusion in specific residues. B: The crystal structure of ORF3a (6XDC) shows the interaction location with TM. C: Molecular docking residues of 6XDC-TM, residues of conventional hydrogen bond (green), residues of carbon-hydrogen bond (cyan), alkyl residues (magenta), pi-alkyl bond (dark magenta), and pi-pi-stacked bound (red).
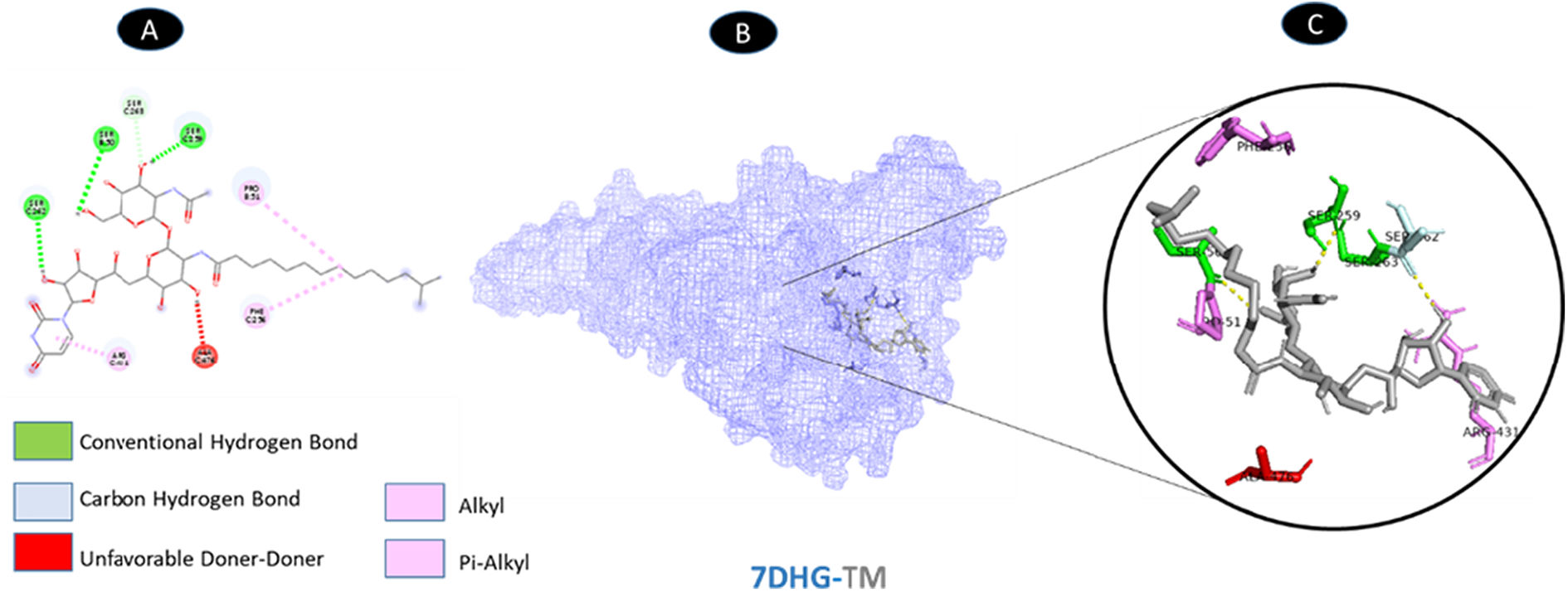
TM and ORF9b have three conventional hydrogen bonding residues (B: SER-50, C: SER-259, and C: SER-262) and one carbon-hydrogen bonding residue (C: SER-263) with eight bounds. There are three weakest alkyl interactions (B: PRO-51, C: PHE-256, and C: ARG-431) as well as one pi-alkyl bound. There is unfavorable donor interaction residue (C: ALA-476). All bonds are illustrated in Fig. 7.
 shows the interaction location with TM. C: Molecular docking residues of 7DHG-TM, residues of conventional hydrogen bond (green), residues of carbon-hydrogen bond (cyan), alkyl, and pi-alkyl residues (magenta), and unfavorable acceptor bond (red).")
Molecular docking of 7DHG-TM. A: 2D interaction shows types of fusion in specific residues. B: The crystal structure of ORF9b (7DHG) shows the interaction location with TM. C: Molecular docking residues of 7DHG-TM, residues of conventional hydrogen bond (green), residues of carbon-hydrogen bond (cyan), alkyl, and pi-alkyl residues (magenta), and unfavorable acceptor bond (red).
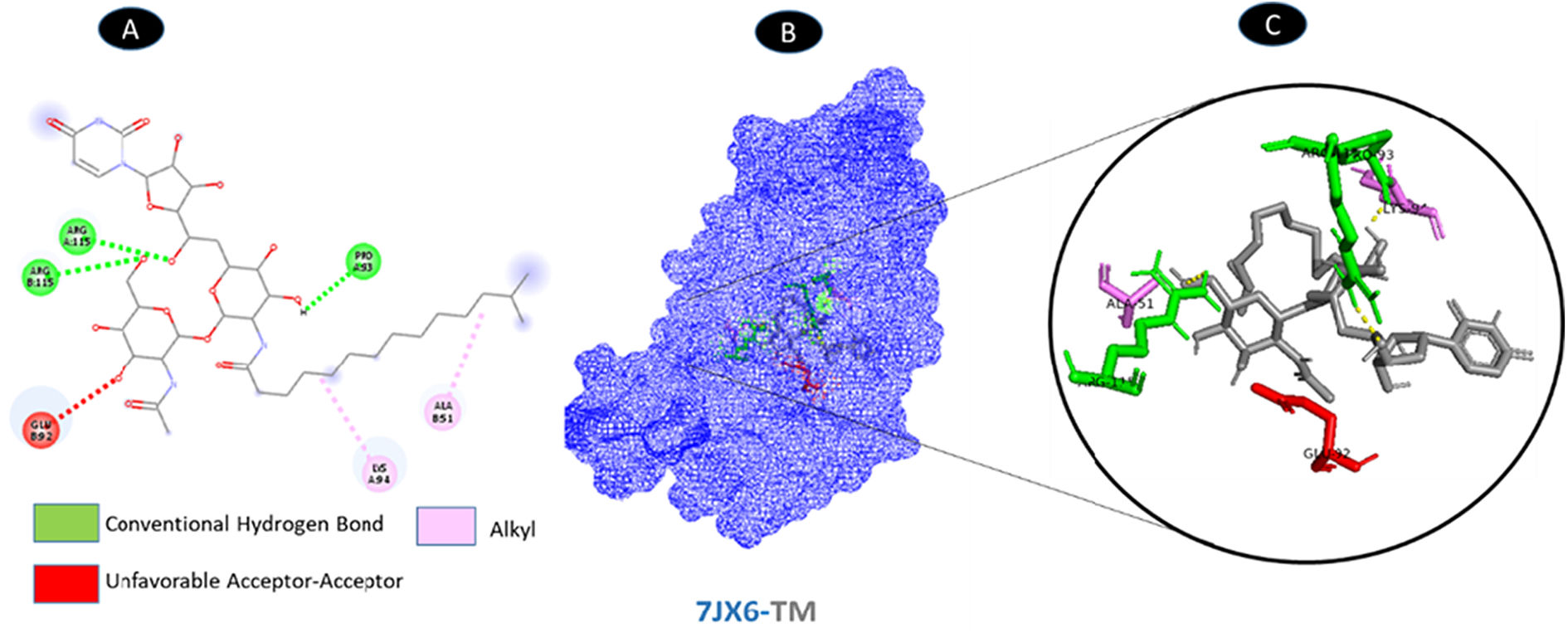
TM and ORF8 have three hydrogen bonding residues in common (A: PRO-93, A: ARG-115, and B: ARG-115). Two probable weakest alkyl connections (A: LYS-94 and B: ALA-51) as well as unfavorable acceptor interaction residue (B: GLU-92). All interactions are illustrated in Fig. 8.
 shows the interaction location with TM. C: Molecular docking residues of 7JX6-TM, residues of conventional hydrogen bond (green), alkyl residues (magenta), and unfavorable donor residue (red).")
Molecular docking of 7JX6-TM. A: 2D interaction shows types of fusion in specific residues. B: The crystal structure of ORF8 (7JX6) shows the interaction location with TM. C: Molecular docking residues of 7JX6-TM, residues of conventional hydrogen bond (green), alkyl residues (magenta), and unfavorable donor residue (red).
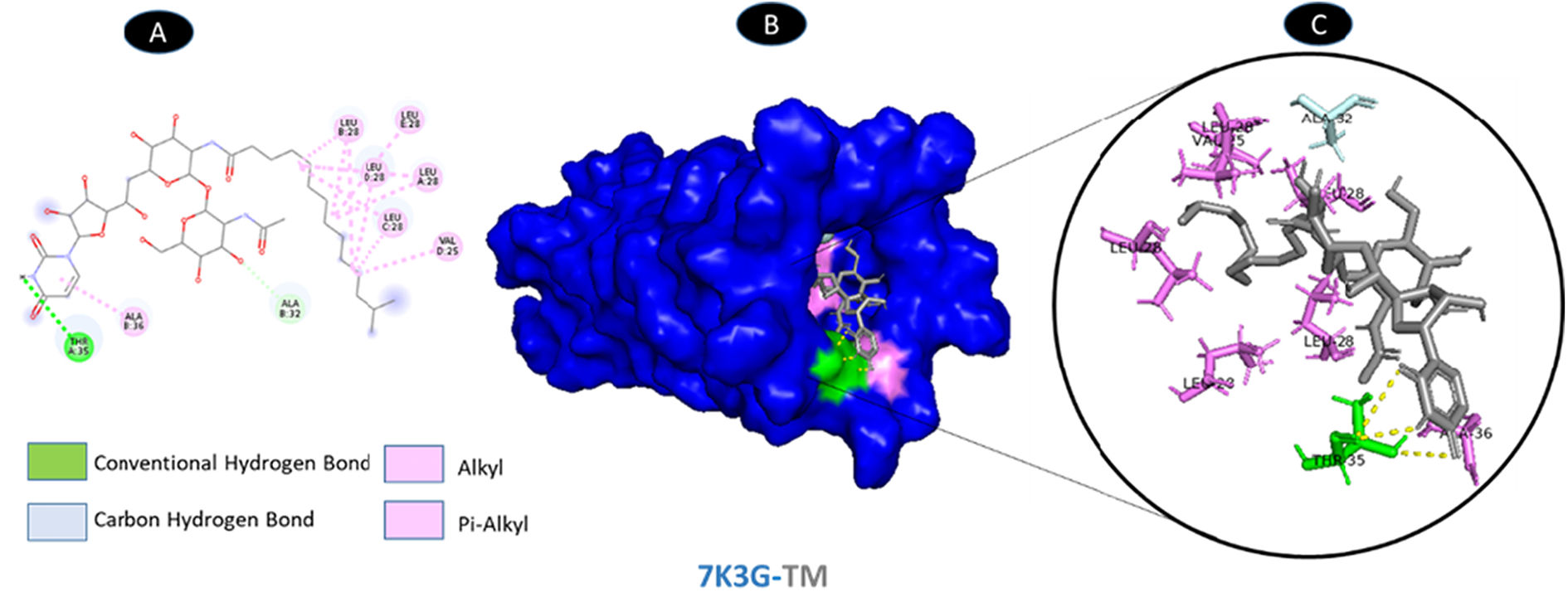
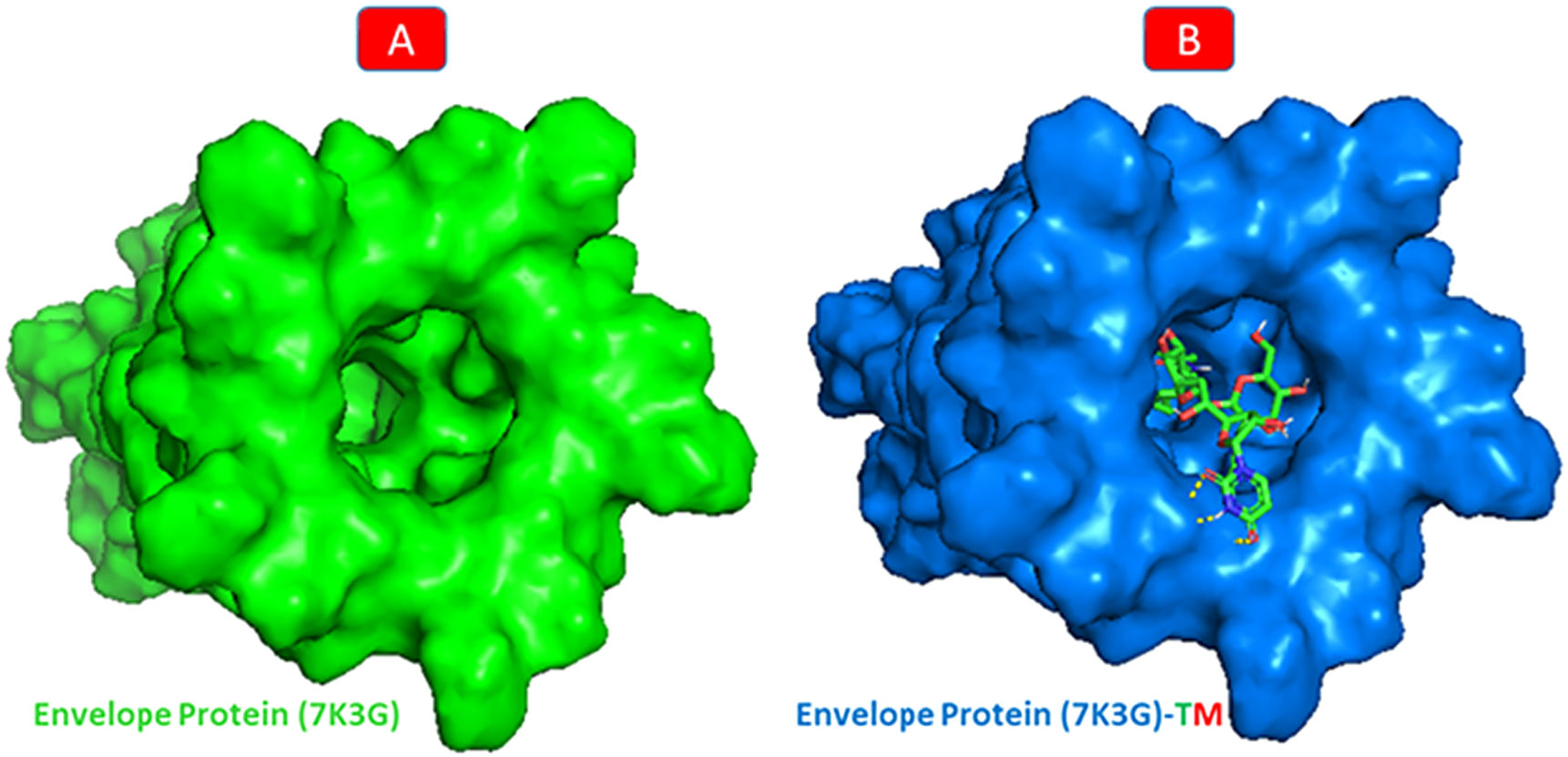
One conventional hydrogen bonding residue (A: THR-35) and a carbon-hydrogen bonding residue (B: ALA-32) with 5 boundaries are shared by TM and envelope protein. As shown in Fig. 9, there are seven probable weakest alkyl and pi-alkyl interactions (A: LEU-28, A: ALA-36, B: LEU-28, C: LEU-28, D: VAL-25, D: LEU-28, and E: LEU-28). Although the docking was in the main pocket of the envelope glycoprotein, there was no change in the structure of the protein after binding to TM, Fig. 10.
 shows the interaction location with TM. C: Molecular docking residues of 7K3G-TM, residues of conventional hydrogen bond (green), residues of carbon-hydrogen bond (cyan), and alkyl and pi-alkyl residues (magenta).")
Molecular docking of 7K3G-TM. A: 2D interaction shows types of fusion in specific residues. B: The crystal structure of envelope protein (7K3G) shows the interaction location with TM. C: Molecular docking residues of 7K3G-TM, residues of conventional hydrogen bond (green), residues of carbon-hydrogen bond (cyan), and alkyl and pi-alkyl residues (magenta).
. B: The molecular docking of 7K3G-TM.")
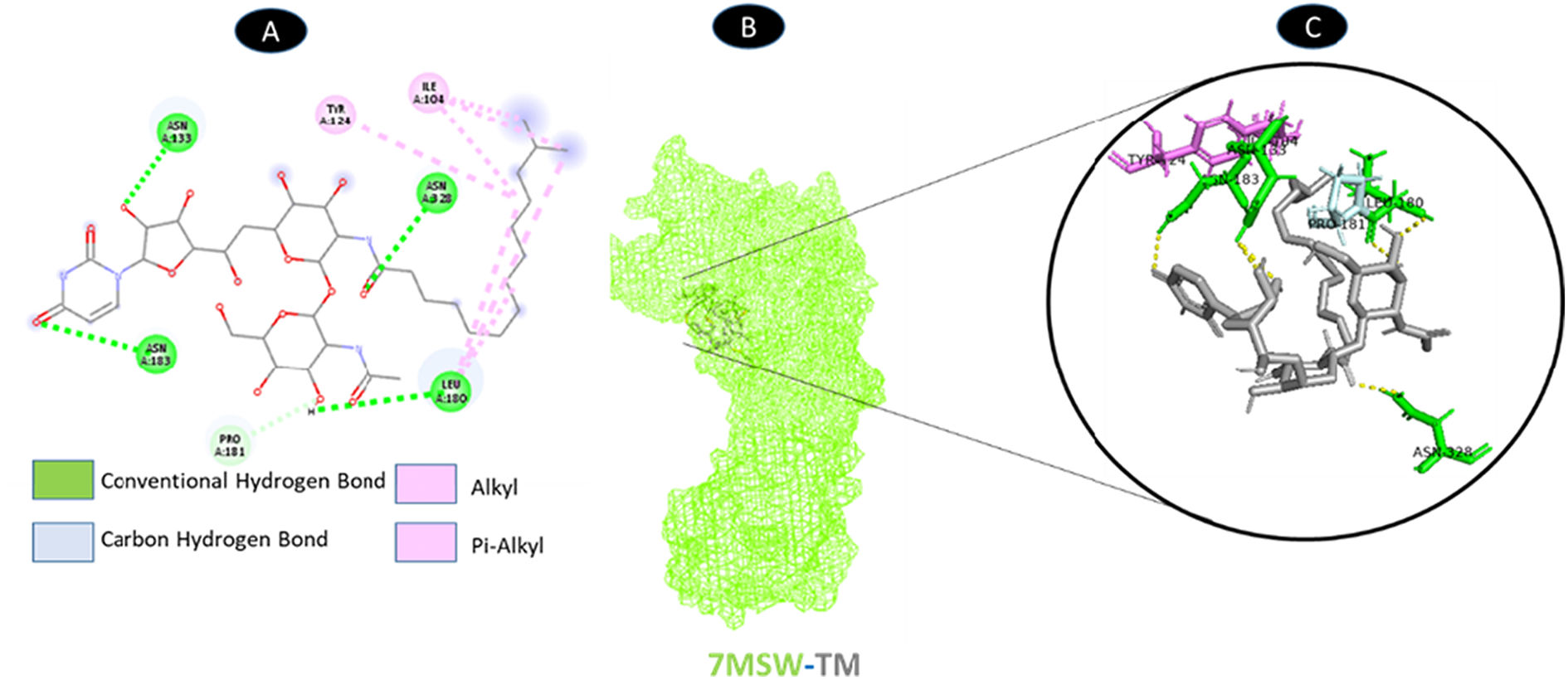
TM and nsp2 share four hydrogen bonding residues (A: ASN-133, A: LEU-180, A: ASN-183, and A: ASN-328), as well as a carbon-hydrogen bonding residue (A: PRO-181) with seven boundaries. As shown in Fig. 11, there are two probable weakest alkyl and pi-alkyl interactions (A: ILE-104 and A: TYR-124).
 shows the interaction location with TM. C: Molecular docking residues of 7MSW-TM, residues of conventional hydrogen bond (green), residues of carbon-hydrogen bond (cyan), and alkyl and pi-alkyl residues (magenta).")
Molecular docking of 7MSW-TM. A: 2D interaction shows types of fusion in specific residues. B: Crystal structure of nsp2 (7MSW) shows the interaction location with TM. C: Molecular docking residues of 7MSW-TM, residues of conventional hydrogen bond (green), residues of carbon-hydrogen bond (cyan), and alkyl and pi-alkyl residues (magenta).
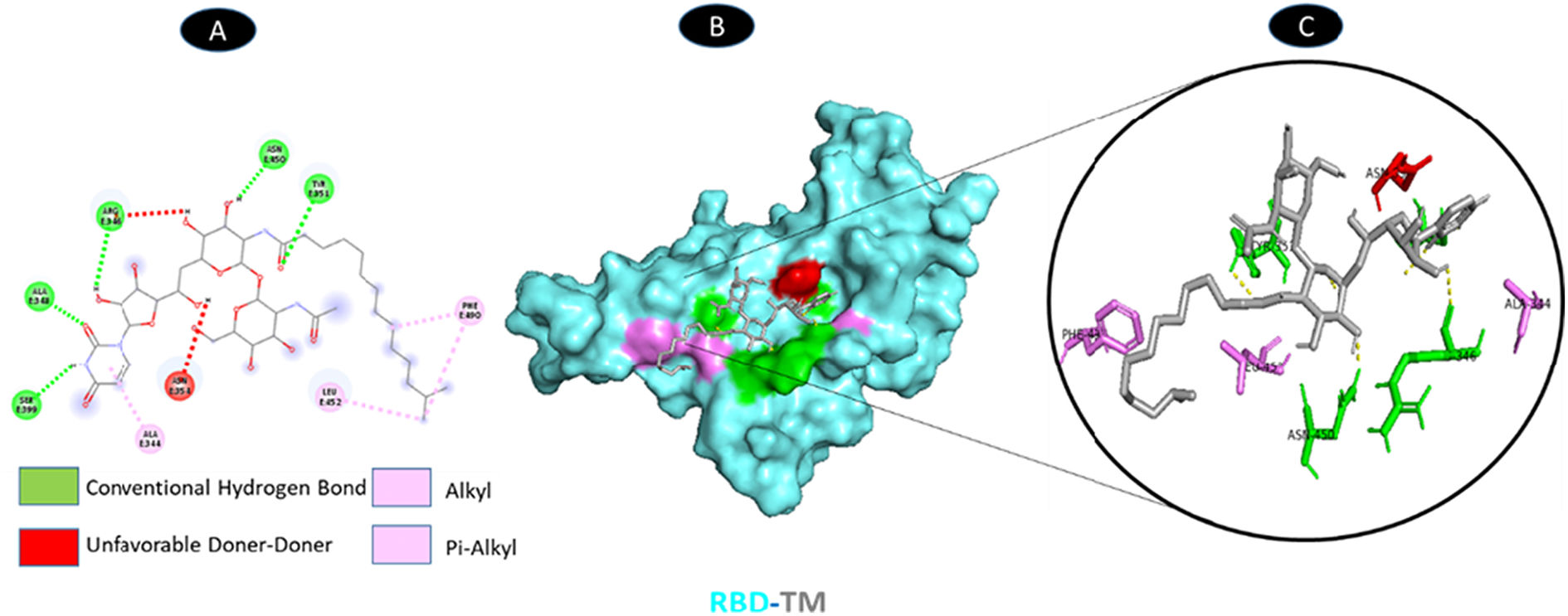
TM and RBD of spike glycoprotein share 5 conventional hydrogen bonding residues (E: ARG-346, E: ALA-348, E: TYR-351, E: SER-399, and E: ASN-450). There is a possible weakest alkyl interactions (E: ALA-344, and E: LEU-452) and one Pi-Alkyl bound residue (E: PHE-490). There are unfavorable donor interaction residues (ASN-354) illustrated in Fig. 12.
-TM. A: 2D interaction shows types of fusion in specific residues. B: Crystal structure of RBD of spike glycoprotein (6M0J) shows the interaction location with TM. C: Molecular docking residues of RBD (6M0J)-TM, residues of conventional hydrogen bond (green), unfavorable donor residue (red), and alkyl and pi-alkyl residues (magenta).")
Molecular docking of RBD (6M0J)-TM. A: 2D interaction shows types of fusion in specific residues. B: Crystal structure of RBD of spike glycoprotein (6M0J) shows the interaction location with TM. C: Molecular docking residues of RBD (6M0J)-TM, residues of conventional hydrogen bond (green), unfavorable donor residue (red), and alkyl and pi-alkyl residues (magenta).
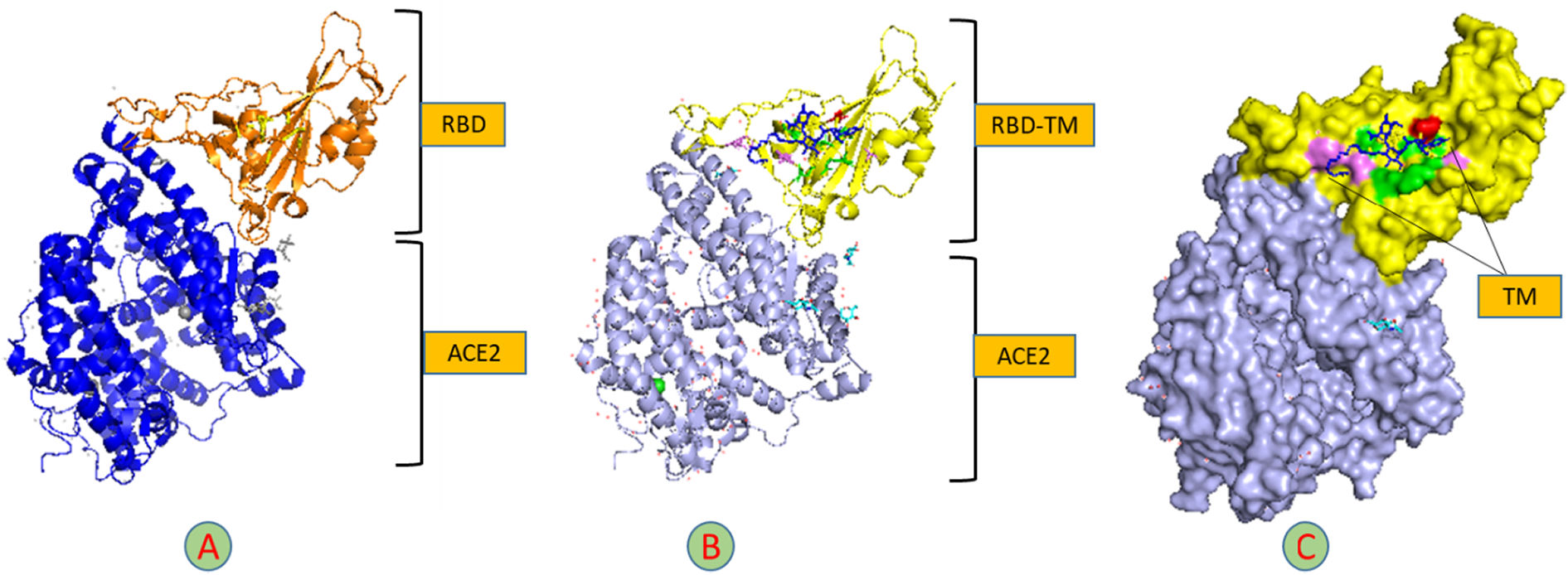
The primary component of the spike glycoprotein through which the virus binds to the Angiotensin-converting enzyme 2 (ACE2) receptor on the host cell is known as RBD. This connection has been established and is identified as ID: 6M0J in the protein data bank. In the current study, we revealed that the tunicamycin binding site in the RBD varies from the ACE2 binding site, which does not effect on the structure of the spike glycoprotein, Fig. 13.
. B: Cartoon structure of the molecular docking of 7K3G-TM. C: The surface structure of 7K3G and TM shows the interaction residues of TM with RBD, hydrogen bond (green), unfavorable donor residue (red), and alkyl and pi-alkyl residues (magenta).")
Molecular docking of TM-RBD-ACE2. A: Cartoon structure of molecular docking of RBD-ACE2 (7K3G). B: Cartoon structure of the molecular docking of 7K3G-TM. C: The surface structure of 7K3G and TM shows the interaction residues of TM with RBD, hydrogen bond (green), unfavorable donor residue (red), and alkyl and pi-alkyl residues (magenta).
Even though a variety of COVID-19 vaccinations are available, the frequency of infections continues to be a major concern.28 On the other side, the rise in the number of deaths drives scientists to look for a drug that may remove or minimize the number of injuries.
Tunicamycin (TM), a nucleoside antibiotic, is a model for drugs that have significant inhibitory effects on protein maturation. On numerous cases, TM has been employed to determine the nonglycosylated protein component of a viral glycoprotein.29,30 TM inhibits subunit 2 of the envelope protein, as well as surface and membrane glycoproteins of coronaviruses, according to a prior study. The glycosylation of coronavirus PTMs like HE and 8ab was decreased by TM. Because TM has been used as an anti-cancer drug and can inhibit coronavirus glycoproteins, we recommend using it to treat SARS-CoV-2.31 Although TM reduces N-linked glycosylation of coronavirus glycoproteins, there is currently no medication that can inhibit O-linked glycosylation. There have been no clinical investigations of TM's effect on tissues or viruses, but our findings may pave the path for TM to be used in clinical trials. Tunicamycin and thapsigargin were shown to decrease cellular stress induced by coronavirus viral and respiratory syncytial virus through distinct pathways for E protein (RSV).32 A study revealed that TM decreased extracellular infectious viral production by almost 99%.33
The PyRx program is known to pick seven models of the protein-ligand binding process as having the highest degree of affinity at various locations. The best three out of seven docking models for each protein were chosen as having the highest binding affinity. When the RMSD equals zero, the optimal fusion model for each protein was picked.
The average root-mean-square distance (RMSD) between the best ranking posture of the tested compounds and their binding pose in the individual crystal structures was found to be less than 2 Å, indicating that the program is capable of docking the compounds properly. The ligand may be isolated, and then docking experiments can be performed to verify the docking procedure.34 The optimal posture is chosen based on binding energy, ligand-receptor interactions, and active site residues once docking is completed. Simply align both docked poses with the co-crystallized structure and calculate the RMSD; a lower number implies that the docking approach is more accurate. Because the RMSD for the selected models for each protein is large, the greatest affinity was chosen when RMSD/up and RMSD/lp equals zero, as shown in table (1). This is what prompted the algorithm to select the first model with a high affinity for the hydrogen and carbon bonding degree between the molecule and the ligand.35,36
There is a high bonding strength with hydrogen and carbon bonds, as well as the existence of weakly linked alkaline bonds, in the proteinase that had the highest affinity for tunicamycin.37,38 The binding of TM to proteinases causes it to be the focus of direct action, altering the enzyme's activity. Although proteases have a high binding affinity, the antagonist may be affected by the presence of three unfavorable acceptor bonds. Similarly, proteins with unfavorable donor or acceptor bonds that impact the degree of fusion (nsp9, ORF9b, ORF8, and RBD of spike glycoprotein) are affected. The ORF7a had the lowest affinity at (-4.9), indicating that TM had no impact on this protein. Similarly, there is a weak binding affinity with TM for (nsp9, ORF8, and RBD).
Binding to TM is a high affinity (-8) for the envelope protein. The pentameric form allows the E protein to dock inside the particular pocket, which is consistent with our earlier research on the binding of doxycycline to the E protein in the same locus where the shape of the protein was altered. Despite the low affinity of the ligation between the TM and the RBD (-6.9), the impact is different because the RBD and ACE2 binding sites are distinct.39
Spike is an essential Coronavirus protein because it connects to the host cell via the ACE2 receptor on the cell membrane's surface. The ACE2 receptor may be found in nearly all organs, although it is especially prevalent in the epithelial cells of the lungs and small intestine. The virus will be unable to enter the host cell if this protein is missing. There has been a lot of study into finding a mechanism to keep the spike protein from being bound to the ACE2, but most of it has failed, and many of them are still being investigated.40,41
In addition to the poor degree of binding and the existence of undesired donor bonds, the current study discovered that the region of TM binding in the RBD had no effect on the region of protein fusion with the ACE2 receptor. Tunicamycin was recommended as a therapy for COVID-19 in prior research.42,43 Because of the strong affinity for interacting with the virus's major proteins, our investigation confirms that this antibiotic should be considered again.
In the current study, it was revealed that the TM may attach to certain regions of the protein, therefore affecting the protein's activity and, as a result, the virion's impact. Depending on this, it's conceivable that the TM inhibits the virus's capacity to infect and, at the very least, keeps it from spreading. This can be accomplished by using it in clinical trials. Previous studies have shown that TM has a powerful effect on carcinoma cells in vitro, so it may have a similar effect on SARS-CoV-2 and suppress it. This study recommends investigating this antibiotic and conducts clinical trials on virus-infected cells. Furthermore, scientists can create effective anti-SARS-CoV-2 drugs using technologies shortly if they work together throughout the world.
ConclusionsTunicamycin has been shown to inhibit proteins during their maturation stage, treat cancer cells by blocking the first step in the G1 phase of the cell cycle, inhibit the production of N-linked glycans in proteins, suppress the production of envelope glycoprotein subunit 2, and inhibit the formation of the spike protein. According to the findings of this investigation, tunicamycin can bind to viral proteins with various degrees of affinity. This antibiotic may affect SARS-CoV-2 in clinical studies.
FundingThis work was done with self-supporting.
Author contributionsDesigning the idea, choosing programs, writing the manuscript, using software, writing the results, analyze and clarifying the purpose of the study were done by the corresponding author.
The author thanks college of Medicine, University of Mosul for documenting this work.