






A new coronavirus strain has wreaked havoc on human lives so the WHO was declared as a pandemic since 20th March 2020. The Membrane glycoprotein MP spans the viral envelope and it has a highly conserved glycosylation sequence.
AimOur study goal was to find out the N-glycosylation, ligand binding sites, and antigenic variations between COVID-19 and other associated viruses.
MethodsWe performed In silico methodologies for serial analysis at both an operational and result/output level is assessed and compared study factors.
ResultsWe detected high similarity in sequence alignment for >89% between COVID-19 MP and other MP of CoVs. Prediction of N-glycosylation and cytotoxic T-cell epitopes, we identified precisely sites between SARS-CoV-2 MP and Pangolin CoV MP 100%. We also didn’t obtain any similarity in ligand binding site residues between MP sequences. Our study didn’t reveal any similarity in CTL epitope predication between coronaviruses under study using the CTLPred server.
ConclusionsOur results exhibit that the membrane glycoprotein of SARS-CoV-2 is closely associated with predecessor SARS-CoVs specifically Pngolin CoV. Prediction of novel CTL epitopes may substantial scopes for the expansion of a peptide-based vaccine for the inhibition virion assembly of SARS-CoV-2.
Una nueva cepa de coronavirus está causando estragos en la humanidad, por lo que la OMS declaró la situación de pandemia el 20 de marzo de 2020. La glicoproteína de membrana MP atraviesa la envoltura viral, y tiene una secuencia de glicosilación altamente conservada.
ObjetivoEl objetivo de nuestro estudio fue averiguar la N-glicosilación, los sitios de unión y las variaciones antigénicas entre COVID-19 y el resto de virus asociados.
MétodosRealizamos metodologías in silico para análisis de series, tanto a nivel operativo como de resultados, y valoramos y comparamos los factores de estudio.
ResultadosDetectamos una gran similitud en cuanto a la alineación de secuencia para > 89% entre la MP de COVID-19 y otras MP de CoV. Prediciendo la N-glicosilación y los epítopos de las células T citotóxicas identificamos con precisión del 100% los sitios entre MP de SARS-CoV-2 y MP de CoV de pangolín. No obtuvimos ninguna similitud en cuanto a los residuos del sitio de unión del ligando entre las secuencias de MP. Nuestro estudio no reveló ninguna similitud en la predicción del epítope CTL entre los coronavirus estudiados, utilizando el servidor CTLPred.
ConclusionesNuestros resultados muestran que la glicoproteína de membrana de SARS-CoV-2 está estrechamente asociada a los SARS-CoV anteriores, específicamente CoV de pangolín. La predicción de los nuevos epítopos CTL puede definir sustancialmente la expansión de una vacuna basada en péptidos para la inhibición del ensamblaje del virión de SARS-CoV-2.
Last December 2019, first reports emerged of a strange illness that was causing mortalities in the Chinese region of Wuhan. This disease is caused by a new strain of coronavirus that generally causes infections of the respiratory tract. As at last of April 2020, the disease had spread to almost every country across all continents of the world, with infections nearing 2,600,000 persons with mortalities hovering around 175,000 individuals. However, the transmission route of SARS-CoV-2 has not been recognized yet, the middling age of the recorded patients was between (55 and 65) years. The male registered >50% of the total. A novel coronavirus infection can stay asymptomatic and may cause mild to severe complications in many cases. Clinical researches have defined diverse symptoms of COVID-19 including fever, dry cough, breath shortness, sneezing, muscle pain as well as conjunctivitis, fatigue, and pneumonia. Some patients might be developed to intense illnesses such as pulmonary edema, acute respiratory distress, renal damage, and multiple failures quickly of organs.1–3 Coronavirus genome ssRNA encodes four main structural proteins that formed the complete virus particles.4
Recently, all detected samples of SARS-CoV-2 have been to be closely allied to Bat-SARS of Betacoronavirus. The length of the encoded viral proteins was originated to be almost similar to Bat-SARS-CoV. A prominent variance was disclosed in the spike protein of SARS-CoV-2 when compared related to other SARS-CoVs.5
The infection is started through the interaction between the viral envelope and host cellular membrane.6 Moreover, the internalization of the virus treasured to the prospect glycosylation sites offered on the viral glycoproteins. The two main viral glycosylated proteins are spike (S) and membrane (M) while the non-glycosylated is the envelope protein (E). The M glycoprotein spans the viral envelope comprised of short N-terminal ectodomain, long C-terminal domain, and the three transmembrane domains.7 Membrane and envelope proteins are coveted for morphogenesis, assembly, and budding of the virion. Glycans are fundamentally concerned with protein post-translational modification and folding to form the N-linkage glycosylation. The main conserved motif composed of Aspargine-X-Serine/Threonine (X: any amino acid except proline). The mannose core binds to the amide nitrogen of asparagine within a conserved motif. The GlcNAc sugar residue is linked via the hydroxyl side chain of Ser and Thr.8 This core of mannose is added in the form of 14 blocked parts in the endoplasmic reticulum and then moves to the Golgi bodies. Glycosylation plays in the fusion activity, antigenic features, and descent of the bioactive modulation on the binding receptors. The glycosylation also plays a substantial role in the status with the host by the effect of the ability of recombinant viruses to proliferate in the hepatic and brain cells.9–11
M protein acts to form a homogeneous complex. Proteins such as spike, nucleocapsid, and envelope can interact with this protein to form heterogeneous complexes. M protein alone cannot induce the formation of virions but it must be activated before the action of E protein before it can be assembled into virions. The all known membrane coronavirus proteins exhibit N and O-linkage glycosylation. The antigenicity of glycosylation is also important concerning the immune synapse formed between T-cell and antigen-presenting cells. Moreover, the glycosylation can potency adjust distinct responses of cellular immunity.11 Our study investigated the topology of M proteins from coronaviridae and identification of the N-glycosylation sites compared between different sources using in silico analysis. We also found out the effect of antigenicity upon predicting the cytotoxic T cell epitopes in M protein.
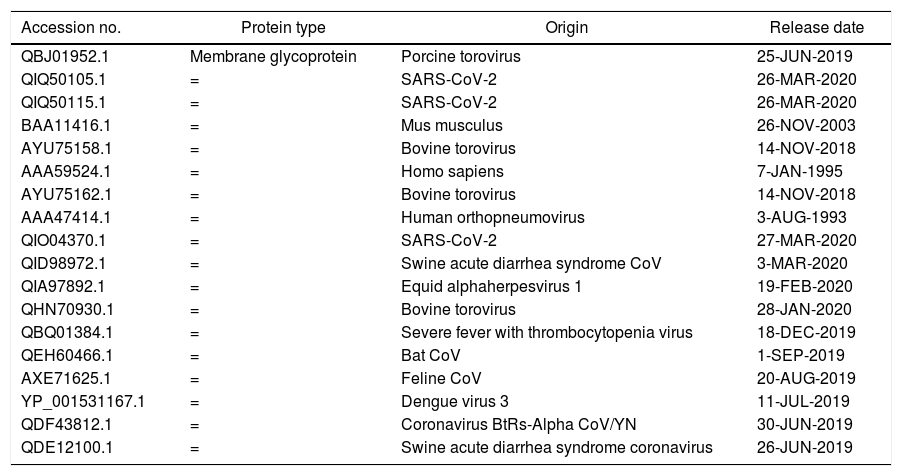
MethodsFor comparison between M glycoprotein of current circulating novel coronavirus with its predecessors and M glycoprotein from different sources, we have selected 18 amino acid sequences from NCBI (Table 1). For analyzing the divergence upon the M glycoprotein sequences, we aligned the collinear complete sequence of MP using CLUSTALW. the phylogenetic tree was generated using CLUSTAL-OMEGA in EMBLE-EBI software and confirmed using the MAFFT version 7 method which is a simple agglomerative hierarchical clustering method (Fig. 1).
Amino acid sequences of M glycoprotein of coronaviruses and from different other sources.
| Accession no. | Protein type | Origin | Release date |
|---|---|---|---|
| QBJ01952.1 | Membrane glycoprotein | Porcine torovirus | 25-JUN-2019 |
| QIQ50105.1 | = | SARS-CoV-2 | 26-MAR-2020 |
| QIQ50115.1 | = | SARS-CoV-2 | 26-MAR-2020 |
| BAA11416.1 | = | Mus musculus | 26-NOV-2003 |
| AYU75158.1 | = | Bovine torovirus | 14-NOV-2018 |
| AAA59524.1 | = | Homo sapiens | 7-JAN-1995 |
| AYU75162.1 | = | Bovine torovirus | 14-NOV-2018 |
| AAA47414.1 | = | Human orthopneumovirus | 3-AUG-1993 |
| QIO04370.1 | = | SARS-CoV-2 | 27-MAR-2020 |
| QID98972.1 | = | Swine acute diarrhea syndrome CoV | 3-MAR-2020 |
| QIA97892.1 | = | Equid alphaherpesvirus 1 | 19-FEB-2020 |
| QHN70930.1 | = | Bovine torovirus | 28-JAN-2020 |
| QBQ01384.1 | = | Severe fever with thrombocytopenia virus | 18-DEC-2019 |
| QEH60466.1 | = | Bat CoV | 1-SEP-2019 |
| AXE71625.1 | = | Feline CoV | 20-AUG-2019 |
| YP_001531167.1 | = | Dengue virus 3 | 11-JUL-2019 |
| QDF43812.1 | = | Coronavirus BtRs-Alpha CoV/YN | 30-JUN-2019 |
| QDE12100.1 | = | Swine acute diarrhea syndrome coronavirus | 26-JUN-2019 |

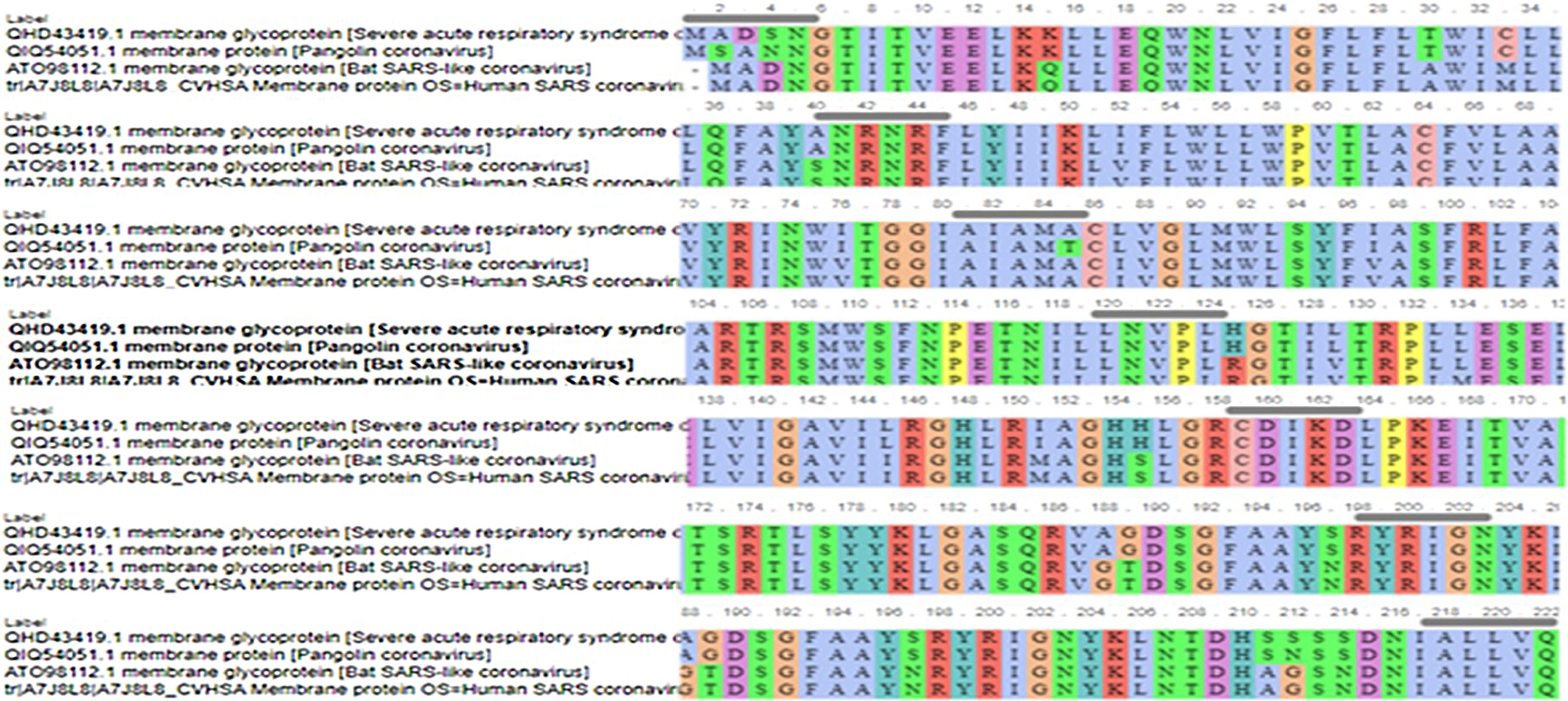
Further selection, we have chosen four closest amino acid sequences in similarity related to coronaviruses for more analysis. MP of SARS-C0V-2 (QHD43419.1), Pangolin CoV (QIQ54051.1), Bat SARS-like CoV (ATO98112.1) and Human SARS-CoV (A7J8L8). We aligned the complete sequences of MP using ViPR (MSA). This data was supported for further testing using T-COFFEE pairwise sequence alignment server. Gaps and mismatches have identified as (–) while the tiny positive scores were detected as (colors other than blue residues), and high scores were identified as blue residues (Fig. 2). The phylogenetic tree was extracted to observe the related sequences (Fig. 3).
Diversity in glycosylation patterns

The glycosylation sites of SARS-CoV-2 MP and related viruses were determined by NetNGlyc 2.0 software to find out differences in the viral attachment sites on the host cell surface (https://www.cbs.dtu.dk/services/NetNGlyc/).12
Antigenic variationThe antigenic variation SARS-CoVs Mp was specified using the NetCTL 1.2 server13 to predict the MHC class I binding peptide, C-terminal cleavage, and transport efficiency of the TAP protein. Cytotoxic T lymphocyte (CTL) epitopes of the MPs detected separately. Then, the results were have compared with a score>0.7. The CTL epitopes were approved by the CTLPred server.
Structural divergence and identified of ligand binding sitesThe protein homology modeling of SARS-CoVs MP sequences was applied using the I-TASSER server to determine the structural divergence between sequences (https://zhanglab.ccmb.med.umich.edu). The generated models of SARS-CoV-2 MP and the other three SARS-CoVs were based on the cryo-EM structure of the SARS-CoVs MP. Based on the local and global RMSD values, the deviation between the structural sequence variations was estimated.14 To detect ligand binding sites for sequences under study, we used I-TASSER and COACH servers.
ResultsPrediction of the phylogenetic tree and sequence alignmentThe phylogenetic tree analysis shows that three sequences of SARS-CoV-2 MP with score identity 100% are closely related with Bat SARS-CoV with score 65% and score (30–40%) with other SARS-CoVs while it was < 20% similarity of MP other sources (Fig. 1). Next multiple sequence alignment and phylogenetic tree-related only SARS-CoVs, the high similarity scores between SARS-CoV-2 MP with Pangolin CoV, Bat SARS-CoV, and Human CoV were 97.75%, 90.5%, and 89.69% respectively (Figs. 2 and 3). This data suggests that MP of SARS-CoV-2 exhibits higher sequence similarity with SARS-CoVs. The sequence variations are desired for interaction with cellular receptors of the minimal binding domains between the amino acids (19–32) and (37–72). The current study set that MP Exhibits 97% identity.
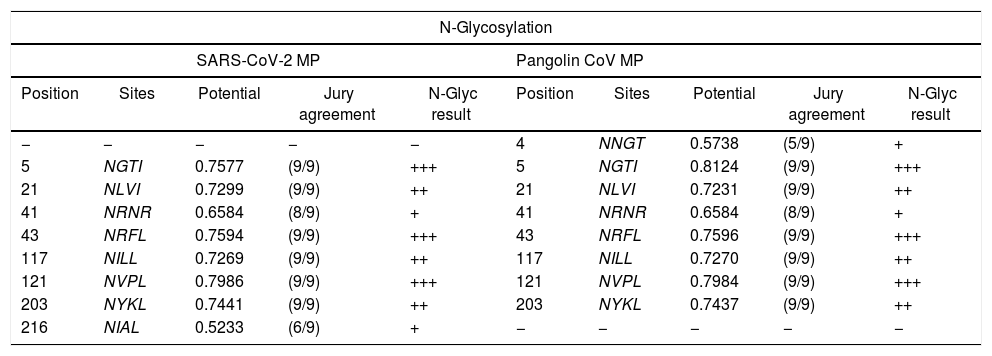
The distinction of N-glycosylation manner for M proteinThe prospect N-linkage residues upon MP under inspection were contrasted and viewed in Table 2. As an emulate with the SARS-CoVs, we have set that MP of SARS-CoV-2 exhibits novel N-glycosylation sites: NGTI, NLVI, NRNR, NRFL, NILL, NVPL, NYKL, and NIAL, that might be the sequence variation results. Besides, we also found that the SARS-CoV-2 MP exhibits mutual N-glycosylation sites offered in SARS-CoVs in gray highlights (Italics text) such as NLVI, NRNR, NRFL, NILL, NVPL, and NYKL (Table 2). Our glycosylation data suggests that the SARS-CoV-2 might interacts with host receptors using linkage residues that could affect the internalization mode and related pathogenesis.
Comparison of N-glycosylation sites between MP of SARS-CoV-2 and 3 sequences of MP SARS-CoVs strains.
| N-Glycosylation | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| SARS-CoV-2 MP | Pangolin CoV MP | ||||||||
| Position | Sites | Potential | Jury agreement | N-Glyc result | Position | Sites | Potential | Jury agreement | N-Glyc result |
| − | − | − | − | − | 4 | NNGT | 0.5738 | (5/9) | + |
| 5 | NGTI | 0.7577 | (9/9) | +++ | 5 | NGTI | 0.8124 | (9/9) | +++ |
| 21 | NLVI | 0.7299 | (9/9) | ++ | 21 | NLVI | 0.7231 | (9/9) | ++ |
| 41 | NRNR | 0.6584 | (8/9) | + | 41 | NRNR | 0.6584 | (8/9) | + |
| 43 | NRFL | 0.7594 | (9/9) | +++ | 43 | NRFL | 0.7596 | (9/9) | +++ |
| 117 | NILL | 0.7269 | (9/9) | ++ | 117 | NILL | 0.7270 | (9/9) | ++ |
| 121 | NVPL | 0.7986 | (9/9) | +++ | 121 | NVPL | 0.7984 | (9/9) | +++ |
| 203 | NYKL | 0.7441 | (9/9) | ++ | 203 | NYKL | 0.7437 | (9/9) | ++ |
| 216 | NIAL | 0.5233 | (6/9) | + | − | − | − | − | − |
| N-Glycosylation | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Bat SARS-CoV MP | Human SARS-CoV MP | ||||||||
| Position | Sites | Potential | Jury agreement | N-Glyc result | Position | Sites | Potential | Jury agreement | N-Glyc result |
| 4 | NGTI | 0.8430 | (9/9) | +++ | 4 | NGTI | 0.8430 | (9/9) | +++ |
| 20 | NLVI | 0.7217 | (9/9) | ++ | 20 | NLVI | 0.7217 | (9/9) | ++ |
| 40 | NRNR | 0.6771 | (8/9) | + | 40 | NRNR | 0.6772 | (8/9) | + |
| 42 | NRFL | 0.7886 | (9/9) | +++ | 42 | NRFL | 0.7887 | (9/9) | +++ |
| 116 | NILL | 0.7213 | (9/9) | ++ | 116 | NILL | 0.7213 | (9/9) | ++ |
| 120 | NVPL | 0.7890 | (9/9) | +++ | 120 | NVPL | 0.7890 | (9/9) | +++ |
| 202 | NYKL | 0.7222 | (9/9) | ++ | 202 | NYKL | 0.7222 | (9/9) | ++ |
| 215 | NIAL | 0.6177 | (8/9) | + | 215 | NIAL | 0.6176 | (8/9) | + |
| 196 | NRYR | 0.7158 | (8/9) | + | 196 | NRYR | 0.7158 | (8/9) | + |
The table shows a comparison of predicted N-glycosylation residues between MP of SARS-CoVs strains. N-glycosylation potential (0.5) was taken as cutoff. The N-glycosylation sites were determined by NetNGlyc 1.0. *Italics indicates the high similarities between N-glycosylation sites between the MP strains. The jury agreement column indicates how many of the nine networks support the prediction.
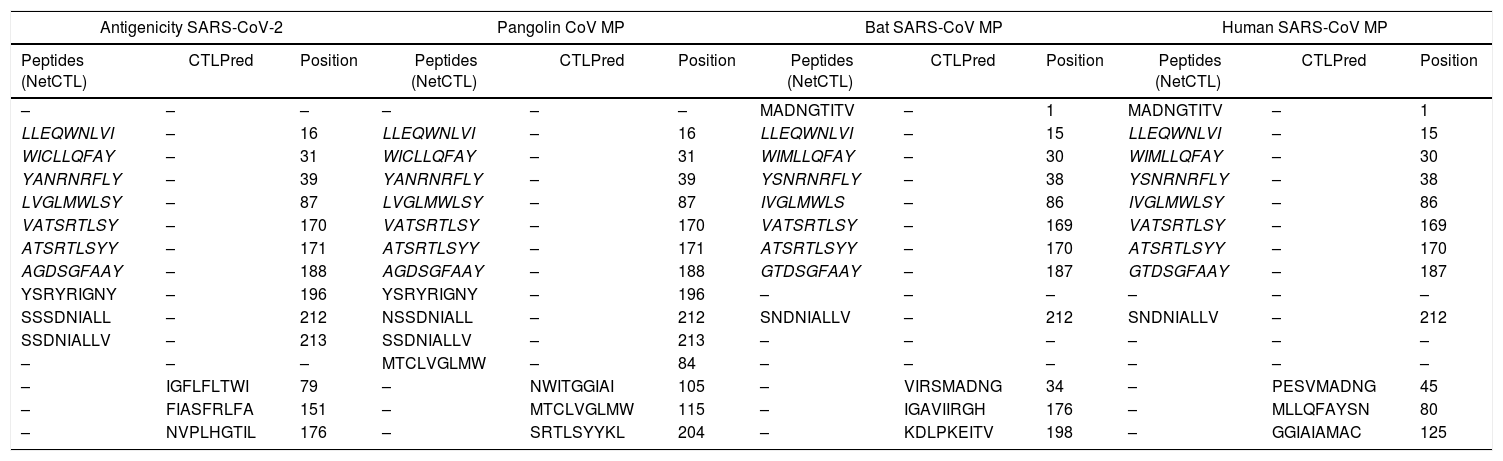
The antigenic diversity of novel and SARS-CoVs MP were matched to define the antigenicity. Our study revealed that most of the CTL epitopes were newly for the SARS-CoV. However, seven epitopes LLEQWNLVI, WICLLQFAY, YANRNRFLY, LVGLMWLSY, VATSRTLSY, ATSRTLSYY, and AGDSGFAAY were found to be identical in the four MP sequences understudy in gray highlight (Italics text) (Table 3). Also, some of the epitopes were set with alternation in single amino acids. The antigenicity data proposed that the SARS-CoV-2 displays altitude antigenic resemblance with related coronavirus and therefore can be theorized as one of the preventive strategies based on a vaccine designed. Furthermore, the predication epitopes may be used to layout effective vaccines. On the contrary, our study recorded that there was no similarity detected in prediction CTL epitopes between SARS-CoV-2 MP and related SARS-CoVs under study using CTLPred tools (Table 3).
Comparison of antigenicity sites between MP of SARS-CoV-2 and 3 sequences of MP SARS-CoVs strains.
| Antigenicity SARS-CoV-2 | Pangolin CoV MP | Bat SARS-CoV MP | Human SARS-CoV MP | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Peptides (NetCTL) | CTLPred | Position | Peptides (NetCTL) | CTLPred | Position | Peptides (NetCTL) | CTLPred | Position | Peptides (NetCTL) | CTLPred | Position |
| – | – | – | – | – | – | MADNGTITV | – | 1 | MADNGTITV | – | 1 |
| LLEQWNLVI | – | 16 | LLEQWNLVI | – | 16 | LLEQWNLVI | – | 15 | LLEQWNLVI | – | 15 |
| WICLLQFAY | – | 31 | WICLLQFAY | – | 31 | WIMLLQFAY | – | 30 | WIMLLQFAY | – | 30 |
| YANRNRFLY | – | 39 | YANRNRFLY | – | 39 | YSNRNRFLY | – | 38 | YSNRNRFLY | – | 38 |
| LVGLMWLSY | – | 87 | LVGLMWLSY | – | 87 | IVGLMWLS | – | 86 | IVGLMWLSY | – | 86 |
| VATSRTLSY | – | 170 | VATSRTLSY | – | 170 | VATSRTLSY | – | 169 | VATSRTLSY | – | 169 |
| ATSRTLSYY | – | 171 | ATSRTLSYY | – | 171 | ATSRTLSYY | – | 170 | ATSRTLSYY | – | 170 |
| AGDSGFAAY | – | 188 | AGDSGFAAY | – | 188 | GTDSGFAAY | – | 187 | GTDSGFAAY | – | 187 |
| YSRYRIGNY | – | 196 | YSRYRIGNY | – | 196 | – | – | – | – | – | – |
| SSSDNIALL | – | 212 | NSSDNIALL | – | 212 | SNDNIALLV | – | 212 | SNDNIALLV | – | 212 |
| SSDNIALLV | – | 213 | SSDNIALLV | – | 213 | – | – | – | – | – | – |
| – | – | – | MTCLVGLMW | – | 84 | – | – | – | – | – | – |
| – | IGFLFLTWI | 79 | – | NWITGGIAI | 105 | – | VIRSMADNG | 34 | – | PESVMADNG | 45 |
| – | FIASFRLFA | 151 | – | MTCLVGLMW | 115 | – | IGAVIIRGH | 176 | – | MLLQFAYSN | 80 |
| – | NVPLHGTIL | 176 | – | SRTLSYYKL | 204 | – | KDLPKEITV | 198 | – | GGIAIAMAC | 125 |
The table shows a comparison of predicted CTL epitopes in MP of SARS-CoV-2 and 3 SARS-CoVs strains. Epitopes were generated by NetCTL 1.0 and confirmed by CTLPred tool where scores > 1 shows highest sensitivity and specificity toward MHC class I. Italics indicates the identical CTL epitopes between 4 strains. There was no similarity of CTL epitope prediction between strains using CTLPred server.
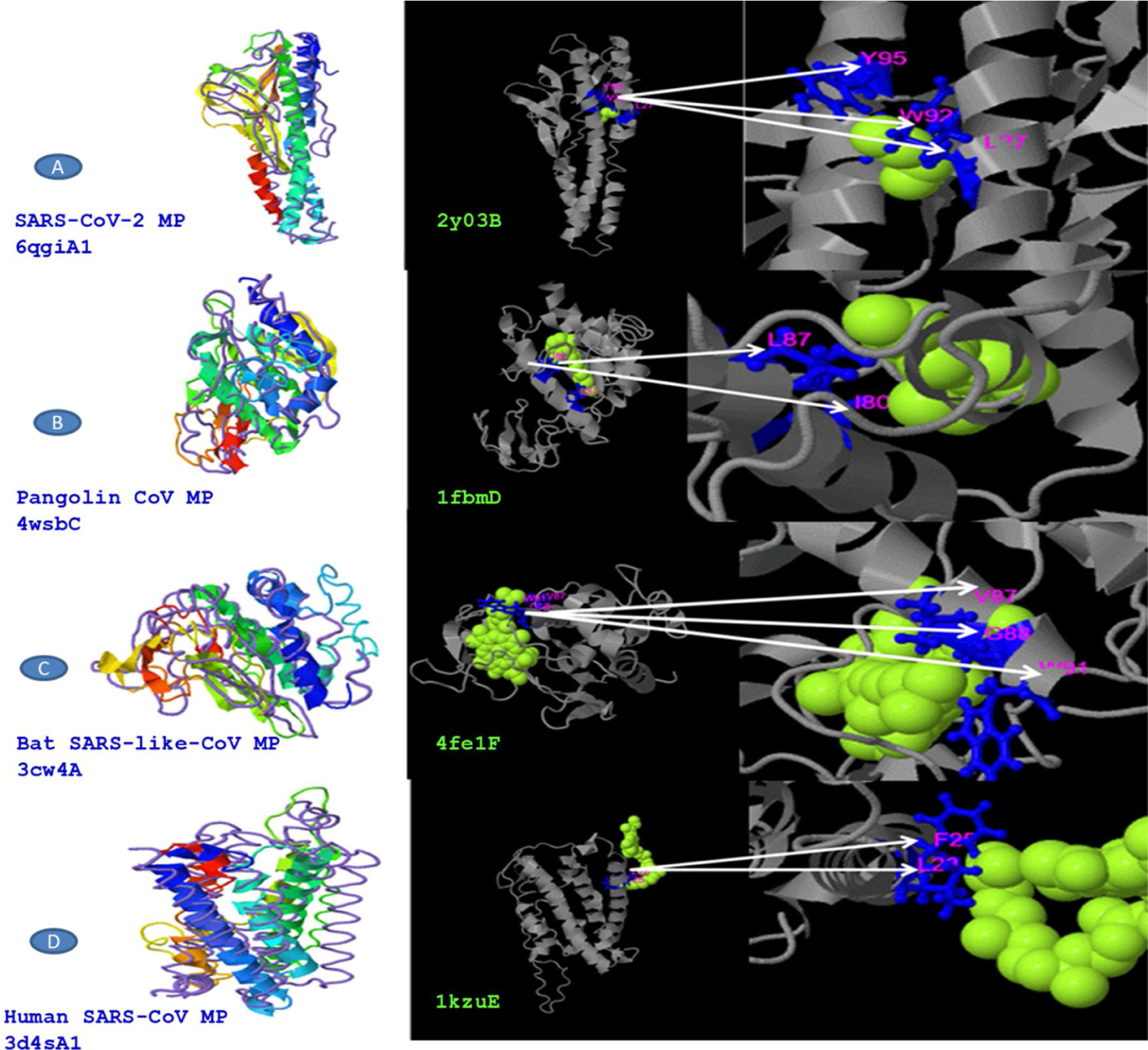
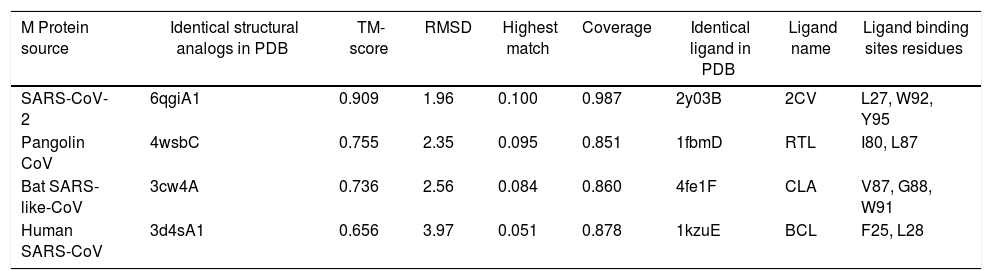
The predicted models were compared for the sequence divergence of MPs. We have found that SARS-CoV-2 MP shows 1.96 angstroms in local RMSD value which scored coverage alignment TM 0.90 with the structure 6qgiA1 in PDB and 0.01 high identity. The ligand-binding site 2y03B in PDB is 2CV (HEGA-10) on the resides (L27, W92, and Y95). The TM-alignment is equal to 0.987 (fig. 4A, Table 4). Furthermore, the 3d structure was identified for MP of Pangolin CoV according to the 4wsbC in PDB. The local RMSD value was recorded at 2.35. The TM- align scored 0.755 with the identity 0.095. The ligand-binding site 1fbmD in PDB is RTL (Retinol) on the resides (I80 and L87). The encasement of the alignment is equal to 0.851 (Fig. 4B, Table 4). The 3d structure was identified for MP of Bat SARS-like-CoV according to the 3cw4A in PDB. The local RMSD value was recorded at 2.56. The TM- align scored 0.736 with the identity 0.084. The ligand-binding site 4fe1F in PDB is CLA (Chlorophylla) on the resides (V87, G88, and W91). The total alignment is equal to 0.86 (Fig. 4C, Table 4). In addition to that, the 3d structure was identified for MP of Human CoV according to the 3d4sA1 in PDB. The local RMSD value was recorded at 3.97. The TM-align scored 0.656 with the identity 0.051. The ligand-binding site 1kzuE in PDB is BCL (Bacteriochlorophylla) on the resides (F25 and L28). The TM-alignment is equal to 0.878 (Fig. 4D, Table 4). Although it has been suggested that the ligand-binding sites are important for vaccine designing, we didn’t obtain any similarity in ligand binding site residues between four MP sequences.

Structural CoVs MP strains close to the target in the PDB and ligand binding sites were predicted in the PBD.
| M Protein source | Identical structural analogs in PDB | TM-score | RMSD | Highest match | Coverage | Identical ligand in PDB | Ligand name | Ligand binding sites residues |
|---|---|---|---|---|---|---|---|---|
| SARS-CoV-2 | 6qgiA1 | 0.909 | 1.96 | 0.100 | 0.987 | 2y03B | 2CV | L27, W92, Y95 |
| Pangolin CoV | 4wsbC | 0.755 | 2.35 | 0.095 | 0.851 | 1fbmD | RTL | I80, L87 |
| Bat SARS-like-CoV | 3cw4A | 0.736 | 2.56 | 0.084 | 0.860 | 4fe1F | CLA | V87, G88, W91 |
| Human SARS-CoV | 3d4sA1 | 0.656 | 3.97 | 0.051 | 0.878 | 1kzuE | BCL | F25, L28 |
I-TASSER uses the TM-align structural alignment in the PDB library. RMSD is the structurally aligned by TM-align. Highest match is the percentage sequence identity in the structurally aligned region. Coverage is equal to the number of structurally aligned residues. COFACTOR infers protein structure comparison. Ligand name is name of possible binding ligand.
The SARS-CoV-2 outbreaks in China have elevated alarm because of its correlated global risk. The administration of SARS-CoV-2 infections depends on the features of the virus including the seriousness of the produced infection, the transmission receptivity, and the availability of vaccines and suitable drugs to control the disease. Most members of coronaviruses are zoonotic which means they can transmit from animals to humans.15,16 The precise origin of SARS-CoV-2 has not been yet identified. The evaluation of SARS-CoV-2 might be arranged into several options including virus virulence, observed new symptoms, conformational changes in the virion, emerging of mutations as well as the assessment of recovery cases after a while.17,18
In this study, we have listed that sequence alignment of amino acids has a considerable variance of less than 20% between membrane protein of coronaviruses and others in different sources. We also detected high similarity for more than 89% between SARS-CoV-2 MP and other MP of CoVs. For instance, a previous study supplied empirical support for the topology of SARS-CoV-2 MP with the N-terminus, long cytosolic C-terminus, and three transmembrane segments.19
We have established novel N-glycosylation residues in the SARS-CoV-2 MP suggesting that viruses may harness different linkages to interact with its receptors. Whilst comparing the N-glycosylation sites, we identified precisely sites between SARS-CoV-2 MP and Pangolin CoV MP 100%. The same similarity was detected between Bat SARS-like-CoV MP and Human CoV MP in all sites with the same score of Jury agreement. From these results, we detected a mutation with the addition of one amino acid, which presented in the reading frame progress with a single codon from the position 20 in Bat SARS-like-CoV to position 21 in SARS-CoV-2 (Table 2). Several shreds of evidence indicated that N and O-glycosylation of the viral envelope proteins are molecular determinants for viral replication and infectivity. Our study agreed with a previous study mentioned that the unique N- and O-linked glycosylation residues of S protein.20–23
Our data showed the sequence similarity, and divergence in the N- glycosylation sites of SARS-C-V-2, MP compared with the SARS-CoVs strain.24,25 We also suggest the range of attachment inhibitors of coronavirus as a treatment option in the current outbreak. Our data may provide a former scale in the structural genomics and labor toward the perception of the development and the mission of the COVID-19.
Our data registered SARS-CoV-2 MP presents unique CTL epitopes that might result in distinct antigenic response related to other SARS-CoVs under study which showed in highlight gray colors: LLEQWNLVI, WICLLQFAY, YANRNRFLY, LVGLMWLSY, VATSRTLSY, ATSRTLSYY, and AGDSGFAAY, although displacement of reading frame one codon in position 15 in Bat SARS-like-CoV MP to position 16 in SARS-CoV-2. Moreover, We revealed that the epitope prediction of SARS-CoV-2 MP is accurately similar to Pangolin CoV MP in highlight gray and pink (Table 3). It has been suggested that CTL epitopes may realize opportunities for the expansion of the vaccine to prevent SARS-CoV-2. However, some epitopes are similar in all glycoproteins.26,27 On the contrary, we didn’t find any similarity in CTL epitope predication between coronaviruses under study using the CTLPred tool (Table 3). It has been suggested that improved detection of CTL and B-cell epitopes may be useful in reverse immunogenetic approaches.28
COACH is a branch in I-TASSER server that joins multiple function surveillance resulted in binding residues. All possible binding ligands and detailed predictions are related to each sequence in PDB. Predication of ligand binding sites might be beneficial for researchers in their decoding of the molecular mechanisms binding in infectivity in addition to the development of antiviral drug or vaccine discovery. From our data, we provided a comparative analysis between the perspective of ligand-binding MP of SARS-CoV-2 and its closest relatives. Unfortunately, we didn’t demeanor any similarity of ligand binding site residues between strains of SARS CoVs. Our previous study conducted that ligand binding sites residues of SARS-CoV-2 spike protein are highly conserved with other S protein relates in SARS CoVs.29
ConclusionsFor the first time, our results demonstrated the emergence of SARS-CoV-2 membrane glycoprotein closely associated with predecessor SARS-CoVs specifically Pangolin CoV. Foremost our data supplies evidence that SARS-CoV-2 MP uses various novel N-glycosylation sites contiguous to SARS-CoVs. Further, the prediction of novel CTL epitopes might sort out chances for the evolution of a predict suitable vaccine to prevent virion assembly of SARS-CoV-2. Furthermore, our disclosure of identical antigenic sites in SARS-CoV-2, Pangolin CoV, Human -CoV, and Bat-SARS-like CoV may suggest the use of specific attachment inhibitors of coronavirus as the actual option of COVID-19 treatment.
Trial registration number (if clinical trial)None.
FundingSelf-support.
Conflicts of interestNo conflict of interest.
The author is very grateful to the college of Medicine, the University of Mosul for improving the quality of this work.