



The development of nonalcoholic fatty liver disease (NAFLD) is strongly associated with the metabolic syndrome as reflected by the fact that approximately 90% of the patients with NAFLD have more than one feature of metabolic syndrome and about 33% have three or more criteria. The physiopathology, epidemiology and therapeutic considerations of the disease are reviewed here. Lipotoxicity plays a predominant role in the pathophysiology of both entities. It leads to accumulation of triglycerides in the liver as a result of an imbalance among the uptake, synthesis, export, and oxidation of fatty acids. Both conditions are very common in Mexico. Using the Adult Treatment Panel diagnostic criteria, the 1994 prevalence of the metabolic syndrome was 26.6%.Although the prevalence of NAFLD is not known, but it can be estimated from the prevalence of obesity (30%). Since NAFLD is found in over two thirds of the obese subjects, this condition may exist in 20% of the adult population. The treatment of both conditions should be based in an integral approach, including the adoption of a healthy lifestyle, weight loss and may be pharmacotherapy. In summary, NAFLD is the hepatic expression of the metabolic syndrome. The study and treatment of these disorders could not be viewed as separate issues.
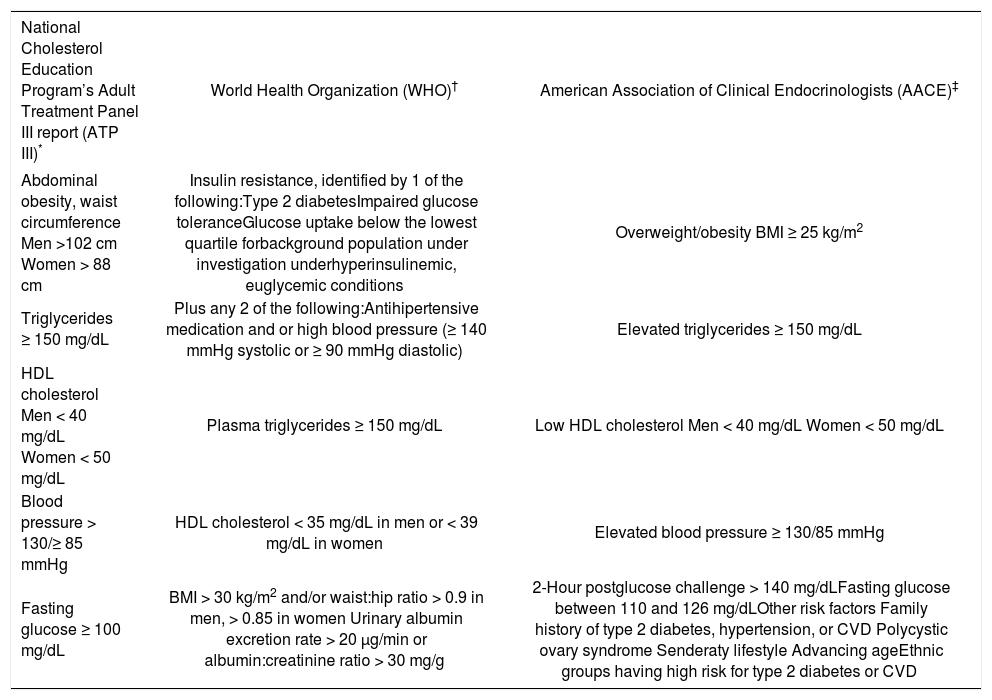
The metabolic syndrome is a condition characterized by a cluster of alterations including glucose intolerance/ insulin resistance, abdominal obesity, atherogenic dyslipidemia (low concentrations of high-density lipoprotein-cholesterol and high concentrations of triglycerides), elevated blood pressure, a proinflammatory and a prothrombotic state. It increases morbidity and mortality, especially due to cardiovascular disease.1,2 On table I the diagnostic criteria for the syndrome according to different organizations are shown.
Diagnostic criteria for the metabolic syndrome.
| National Cholesterol Education Program’s Adult Treatment Panel III report (ATP III)* | World Health Organization (WHO)† | American Association of Clinical Endocrinologists (AACE)‡ |
|---|---|---|
| Abdominal obesity, waist circumference Men >102 cm Women > 88 cm | Insulin resistance, identified by 1 of the following:Type 2 diabetesImpaired glucose toleranceGlucose uptake below the lowest quartile forbackground population under investigation underhyperinsulinemic, euglycemic conditions | Overweight/obesity BMI ≥ 25 kg/m2 |
| Triglycerides ≥ 150 mg/dL | Plus any 2 of the following:Antihipertensive medication and or high blood pressure (≥ 140 mmHg systolic or ≥ 90 mmHg diastolic) | Elevated triglycerides ≥ 150 mg/dL |
| HDL cholesterol Men < 40 mg/dL Women < 50 mg/dL | Plasma triglycerides ≥ 150 mg/dL | Low HDL cholesterol Men < 40 mg/dL Women < 50 mg/dL |
| Blood pressure > 130/≥ 85 mmHg | HDL cholesterol < 35 mg/dL in men or < 39 mg/dL in women | Elevated blood pressure ≥ 130/85 mmHg |
| Fasting glucose ≥ 100 mg/dL | BMI > 30 kg/m2 and/or waist:hip ratio > 0.9 in men, > 0.85 in women Urinary albumin excretion rate > 20 μg/min or albumin:creatinine ratio > 30 mg/g | 2-Hour postglucose challenge > 140 mg/dLFasting glucose between 110 and 126 mg/dLOther risk factors Family history of type 2 diabetes, hypertension, or CVD Polycystic ovary syndrome Senderaty lifestyle Advancing ageEthnic groups having high risk for type 2 diabetes or CVD |
According to some groups, insulin resistance is the main feature of the metabolic syndrome. Insulin resistance refers to a condition in which a greater amount of insulin are required to elicit a normal biologic response at a cellular, organ or whole-body level. As insulin resistance increases, a greater degree of compensatory hyper-insulinemia is present.1,3
In Mexico there is a high prevalence of the metabolic syndrome. Using the Adult Treatment Panel (ATP III) diagnostic criteria, a prevalence of 26.6% was estimated and according to the WHO criteria the prevalence was 13.61%. A significant proportion of the total cases (35.2%) are less than 40 years of age and in people without diabetes the age distribution is shifted to a younger group regardless the diagnostic criteria used. The metabolic syndrome was more common in women than in men (13.39% vs 13.79%) according to the WHO criteria and more common in men (28.5% vs 25.2%) according to the ATP III. The number of cases was significantly lower with the WHO definition; in fact only 43.4% of the subjects who fulfilled the ATP III criteria were diagnosed using the WHO definition.4
Regardless of its definition, the metabolic syndrome is an important public health concern due to its high prevalence and because its association with an increase in morbidity and mortality mainly due to cardiovascular disease.
Nonalcoholic fatty liver diseaseNonalcoholic fatty liver disease (NAFLD) refers to a histological spectrum of liver damage from simple steatosis to advanced fibrosis and cirrhosis in individuals without a relevant alcohol consumption (< 20 g/day).5 It has been recognized as a major cause of liver-related morbidity and mortality.6 Nonalcoholic steatohepatitis (NASH) is believed to be an intermediate stage in the progression from steatosis to cirrhosis. Histological changes of NASH are characterized by steatosis, mixed inflammatory cell infiltration, necrosis and progressive fibrosis, ultimately leading to cirrhosis and end-stage liver disease. These are similar to alcohol-induced hepatitis. The probability of developing advanced hepatic fibrosis is significantly greater in individuals with steatohepatitis than in those with simple steatosis.7,8 In most cases, fatty liver does not progress to end-stage liver disease, but approximately 20% to 30% of cases with NAFLD have histological signs of fibrosis, inflammation and necrosis, indicating the presence of NASH. These patients are at higher risk of developing cirrhosis, terminal liver failure and hepatocellular carcinoma.9
In Mexico the prevalence of NAFLD is not known, but it can be estimated from the prevalence of obesity and type 2 diabetes mellitus. The 2006 National Survey of Health and Nutrition in Mexico found that nearly 30 percent of the population (34.5% of women and 24.2% of men) is obese. Estimations consider that steatosis is found in over two thirds of the obese population, meaning that around 20% of the population may have NAFLD. In addition, according to the Survey the prevalence of diabetes mellitus in the adult population was 7% and NAFLD is found in about 50% of patients with diabetes.10
It has been estimated that high proportions of the cryptogenic cirrhosis cases have the clinical features and are related to NAFLD.
Pathophysiology of the metabolic syndromeInsulin resistance is a key event in the pathophysiology of the metabolic syndrome. Hyperinsulinemia, caused by an increased insulin secretion by the pancreatic beta cells and decreased insulin degradation by the liver, is a compensatory phenomenon to insulin resistance. Hyper-insulinemia leads to an increase in fat mass, lipogenesis, and is associated with increased concentrations of free fatty acids (FFA). These changes are linked with further reduction in insulin signaling and an increase in both hepatic glucose and lipid production.11
A disruption in lipid metabolism has been implicated in the pathogenesis of insulin resistance. FFA inhibits carbohydrate oxidation, insulin-stimulated glucose uptake and its incorporation into glycogen due to a decrease in the activity of glycogen synthase.12,13 Also, a disturbance in fatty acid partitioning could contribute to insulin resistance due to the inhibition of cytosolic long-chain fatty (LCFA) acyl CoA oxidation.14,15 The mechanism involves malonyl-CoA, which inhibits carnitine palmitoyltransferase (CPT). CPT controls the transfer of LCFA acyl-CoA from the cytosol into the mitochondria where they are oxidized.3 There is an association between elevations of malonyl-CoA and insulin resistance. An increase in its concentration leads to an increase in cytosolic LCFA acyl CoAs, its esterification, formation of diacylglycerol, triglycerides, ceramide, and reactive oxygen species, all linked to insulin resistance.14,16,17
The excess of nonesterified fatty acids and the increased intracellular lipid content correlates with the presence of insulin resistance.18 The adipocyte is as a reservoir of fuel stored as triglycerides during caloric abundance for subsequent release during periods of caloric need. When the adipocyte no longer functions as a store for lipids, fatty acids are deposited as triglycerides in ectopic sites such as muscle, liver and visceral fat. Abnormal accumulation of fat in the muscle and other tissues plays an important role in the etiology of insulin resistance.15,19
The adipocyte, in addition to its role as a reservoir of fuel, is an active endocrine organ that produces and secretes a number of cytokines (adipokines) that contribute to the regulation of metabolic processes. Insulin resistance states are characterized by elevated expression and production of adipokines, among the most relevant proinflammatory cytokines secreted by the adipose tissue are tumor necrosis factor-alpha (TNF-a), resistin and plasminogen activator inhibitor (PAI), which contribute to the alterations that lead to insulin resistance.20
Adiponectin is produced exclusively by adipose tissue and it has been linked to insulin resistance in humans. Its concentrations are decreased in patients with obesity, insulin resistance, type 2 diabetes and NAFLD.6 It has been demonstrated that adiponectin decreases insulin resistance by increasing fatty-acid oxidation, reducing triglyceride content in skeletal muscle and liver and suppressing hepatic glucose production.21
Adiponectin activates AMPK (a fuel sensoring enzime) and stimulates AMPK-mediated events such glucose transport and fatty acid oxidation in muscle.21,22 TNF-a has a role antagonizing the effects of adiponectin, contributing to insulin resistance. Furthermore, some data suggest that adiponectin may have a protective role in liver injury in alcoholic and nonalcoholic fatty liver disease in mice due to its antagonistic effects against TNF-a.23
Summarizing, the systemic effects of decreased insulin sensitivity reflect the lipotoxic effects of fatty acids as well as an imbalance of the cytokines produced by adipose tissue.
Endothelial dysfunction is another feature that has been observed in humans with the metabolic syndrome. Endothelial dysfunction is thought to be one component of the inflammatory process that initiates atherogenesis. It is manifested as an impaired endothelium-dependent relaxation and increases in circulating adhesion molecules like vascular cell adhesion molecule (VCAM) 1, intracellular adhesion molecule (ICAM) 1, and E-selectin. E-selectin is induced by inflammatory cytokines, whereas ICAM-1 and VCAM-1 are expressed by endothelial cells in response to inflammatory cytokines, elevated levels of FFA and oxidized low-density lipoprotein cholesterol.24,25
There is a clear association between hepatic lipid deposition and several features of insulin resistance (hyperinsulinemia, hypertriglyceridemia and low HDL concentration) and less hepatic insulin sensitivity is associated with more liver adipose tissue content.26 The molecular mechanisms associated with the development of hepatic insulin resistance have been studied and include a decrease in insulin-stimulated tyrosine phosphorylation of IRS-1 and IRS-2, which blocks the ability of insulin to activate glycogen synthase, diminishes the ability to store glycogen and increases gluconeogenesis.27
The increases in intracellular triglyceride content in tissues of humans with insulin resistance and metabolic syndrome can be related to increases in the uptake of FFA from plasma, an enhanced rate of de novo fatty acid synthesis, and a dysregulation of intracellular lipid partitioning in which fatty acid oxidation is impaired and its esterification enhanced. The metabolic and inflammatory changes observed in the liver of patients with nonalcoholic fatty liver disease resemble those of lipotoxicity in other organs.16
Insulin resistance is usually compensated by islet beta-cells adaptation with both increased beta-cell number and increased beta-cell function. Both abnormally high glucose and lipids have been implicated in the failure of beta-cells. Postprandial hyperglycemia and dyslipidemia are features that occur before the development of insulin secretory defect. Increases in plasma FFA increase short-term insulin secretion whereas prolonged increases in the concentration of saturated fatty acids and glucose together cause dysfunction and damage to the beta cell resulting in apoptosis.28
The increase in free FFA from adipocyte lypolisis or from excess dietary intake activates protein kinase C (PKC), which phosphorylates serine residues on the insulin receptor and insulin receptor substrates (IRS), impairing tyrosine phosphorylation required for normal signaling to increase glucose transport, glycogen synthesis and other insulin-stimulated events. In the liver the ability of insulin to inhibit gluconeogenesis and glycogenolysis is impaired; all these events increase hepatic insulin resistance which led to elevated glucose production and hyperglycemia.16 Hyperinsulinemia induces overexpression SREBP1-c leading to increased hepatic lipogenesis. The liver responds increasing cholesteryl ester synthesis, production of very low density lipoproteins and synthesis of triglycerides exacerbating dyslipidemia.29
The pathophysiology of the metabolic syndrome is complex; the evidence points to insulin resistance as a central factor in the initial disturbance and its final manifestations.
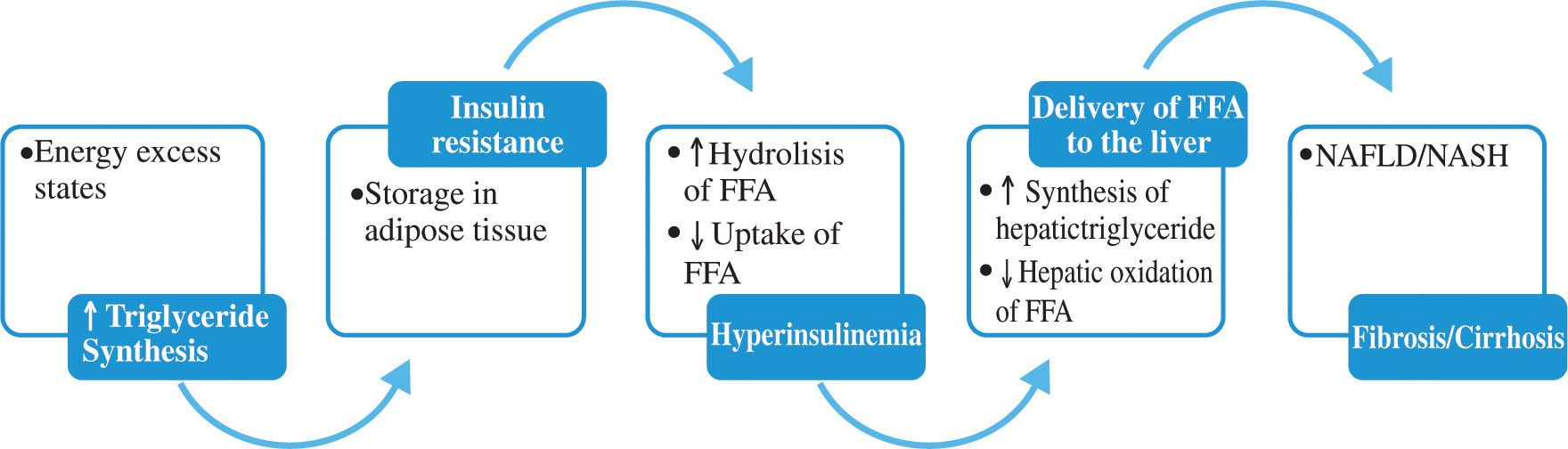
Pathophysiology of the non-alcoholic fatty liver diseaseTraditionally the pathogenesis of NAFLD has been proposed related first to factors that lead to hepatic steatosis and next to “a second hit” that leads to hepatic damage progression. The development of NAFLD requires the accumulation of triglycerides within the hepatocytes. The triglycerides, integral components of lipoprotein particles synthesized by the liver and small intestine, are a source of stored energy in adipose tissue and skeletal muscle. Dietary FFA and released from adipose depots are used for hepatic triglyceride synthesis. In the hepatocyte they undergo oxidation or they become esterified into triglycerides. A: diacylglycerol acyltransferase (DGAT) is the enzyme that catalyzes de final step in triglyceride synthesis; ultimately triglycerides exit the liver as VLDL.30
This accumulation of triglycerides in hepatic and other tissues results from an imbalance among the uptake, synthesis, export, and oxidation of fatty acids.11 In states of energy excess, hepatic DGAT activity and triglyceride synthesis are increased. Triglycerides are exported from the liver in lipoprotein particles and derived to extrahepatic tissues.30
Within adipose tissue, lipoprotein lipase hydrolyzes liver-derived triglyceride and liberates FFA which are transported into adipocytes. The FFA derived from visceral adipocytes, reach the liver through the portal vein, overload hepatocytes with lipid and promote hepatic triglyceride storage. In insulin resistant states there is an increase in lipolysis in adipose depots and inhibition of FFA uptake, increasing FFA delivery to the liver. On figure 1 the mechanisms of hepatic fat deposition are illustrated.29,30

Triglyceride synthesis and NAFLD.
Triglyceride synthesis increases in states of energy excess. Insulin resistance and hyperinsulinemia lead to increase lipolysis of triglyceride depots in adipose tissue, amplifying the deriver of FFA to the liver. Insulin further stimulates liver triglyceride synthesis while inhibiting fatty acid oxidation inhibiting production of VLDL.
Studies in animal models demonstrated improvements in hepatic steatosis when triglyceride synthesis was inhibited. Interestingly, in a recent animal model, synthesis of triglycerides was a protective mechanism limiting the progression of NAFLD to NASH and fibrosis. The authors hypothesize that triglyceride accumulation is a hallmark of livers that are more exposed to FFA and hepatic VLDL synthesis is a protective mechanism.30
Alteration in the secretion of adipocytokines from adipocytes contributes to metabolic and inflammatory abnormalities such as alteration in the rate of the synthesis of triglycerides in the hepatocytes and increased lipolysis in central adipose tissue. As mentioned before, low circulating levels of adiponectin have been linked to several components of the metabolic syndrome. In the liver, adiponectin increases insulin sensitivity by decreasing the expression of hepatic gluconeogenic enzymes (phosphoenolpyruvate carboxykinase and glucose-6-phosphatase) and the rate of endogenous glucose production. In addition, adiponectin suppresses lipogenesis and activates free fatty acid oxidation.31,32
Tumor necrosis factor a, an adipocyte-derived cytokine, is an important mediator of insulin resistance, it impairs insulin signaling by decreasing the tyrosine kinase activity of the insulin receptor (IR). Through serine residues phosphorylation in the IRS-1, reduces the capacity of IRS-1 to be phosphorylated by the IR and induces a state of cellular insulin resistance.33 It has been proposed that overproduction of TNF-a plays a key role in the pathogenesis of fatty liver; by increasing mitochondrial generation of reactive oxygen species, promotes hepatocyte apoptosis and recruits inflammatory cells to the liver.30 In ob/ob mice, a model for NAFLD, fatty liver disease is significantly improved by inhibition of TNF-a production.8
Leptin is also produced by adipose tissue in proportion to adipose tissue mass, when leptin action is lacking, whether a consequence of its deficiency or due to leptin resistance, generalized steatosis develops in liver, heart, pancreatic islets, kidneys and skeletal muscle. Leptin might function as an antisteatotic hormone aimed at preventing free fatty acid entry and fat accumulation in non-adipose tissues.34 Additionally, leptin appears to be essential for developing fibrosis in response to chronic liver injury due to induction of transforming growth factor /3 1.35
Once hepatic steatosis is present other factors promote progression to NASH. Patients at risk for more severe disease are those with several features of the metabolic syndrome, indicating that insulin resistance might play a role in disease progression.11 Oxidative stress has been recognized as a mechanism responsible for progression of liver damage. The oxidative stress includes reactive oxygen species (ROS) production and lipid peroxidation eventually leading to NASH. The potential sources for the ROS include hepatic cytochrome P450 2E1, mitochondria and iron overload.7 Mitochondrial damage also produce ROS which may trigger steatohepatitis and fibrosis by lipid peroxidation, cytokine induction and induction of Fas ligand.5 Excessive fatty acid oxidation is a major contributor to oxidative stress in the liver. Hepatic stellate cells are activated during chronic liver injury and produce excessive extracellular matrix leading to fibrosis; they also produce ROS contributing even more to the oxidative stress.7
Linkage of the metabolic syndrome to non-alcoholic fatty liver diseaseNAFLD can be considered as the hepatic representation of the metabolic syndrome.29 The development of NAFLD is strongly associated with the metabolic syndrome as reflected by the fact that approximately 90% of the patients with NAFLD have more than one feature of metabolic syndrome and about 33% have three or more criteria.36 Furthermore, with the addition of each of the components of the metabolic syndrome the risk of steatosis increases exponentially.37
Nonalcoholic fatty liver disease has been consistently associated with obesity (60-95%), type 2 diabetes mellitus (28-55%) and dyslipidemia (27-92%).11
The presence of the metabolic syndrome is associated with a potentially progressive and severe liver disease. In addition, the likelihood of developing NASH increases with the severity of obesity.11,36 There is a universal association between NASH an insulin resistance regardless of body mass index, suggesting that insulin resistance is a central factor in the pathogenesis of NASH.38
The elevated expression of TNF-a in the liver seen in NAFLD can represent a link between the development of insulin resistance and hepatic steatosis. TNF-a has been proposed as an important mechanism of peripheral insulin resistance in obesity and type 2 diabetes. It is liked to increased oxidative stress and cell death in the liver, potentiating the development of liver fibrosis and progression to NASH.7
Therapeutic considerationsThe diagnosis and treatment of the metabolic syndrome is a public health problem. The syndrome is associated with an increased morbidity and mortality mainly due to cardiovascular disease. The treatment of patients with NAFLD should include the identification and treatment of the associated metabolic abnormalities to ameliorate the cardiovascular risk and to improve NALFD.
Many therapeutic options such as diet and exercise are often difficult to maintain successfully. A primary target of intervention for metabolic syndrome should be weight reduction and the prevention of obesity. Long-term maintenance of weight loss is best achieved when regular physical activity is included in the weight-reduction regimen. Weight reduction has been associated with improvement in liver function tests and in histological findings related to fatty liver.39 The rate of weight loss should be gradual, because in patients with a high degree of fatty infiltration, rapid weight loss may promote inflammatory hepatitis and fibrosis due to an increase in adipose tissuelipolysis and the release of FFA contributing to the exacerbation of preexistent liver steatosis, at least in some patients. A target of weight loss of about 10% of body weight over six months seems reasonable.29,40-42 The standard exercise recommendation is a daily minimum of 30 minutes of moderate intensity physical activity.43
There is no established drug treatment for steatohepatitis. Multiple pharmacologic interventions have been attempted with variable success including pentoxifilline, orlistat, vitamin E, ursodeoxycholic acid and lipid-lowering agents. Further randomized, well controlled trials are required to determine the efficacy of these drugs. There are two classes of drugs currently available that reduce insulin resistance. These are metformin and insulin sensitizing drugs (thiazolidinediones). Long term treatment with metformin significantly reduced transaminase concentrations and liver size.44 Thiazolidinediones have been shown to improve insulin sensitivity in patients with type 2 diabetes, they decrease serum aminotransferase levels, decrease liver fat, and improve the histological features of steatohepatitis (steatosis, ballooning necrosis, and centrilobular inflammation).45
The control of serum glucose and lipids levels should be part of the management. Lipid lowering agents like statins and fibrates are useful for the treatment of dyslipidemia and may improve liver enzymes and hepatic inflammation.42
ConclusionsNon alcoholic fatty liver disease is a manifestation of the metabolic syndrome. The physiopathology reviewed in this article focused on the lipotoxicity associated with metabolic syndrome and considers NAFLD as a final expression of this abnormality that can have significant liver morbidity and mortality. The treatment of both conditions as with all the metabolic disturbances that characterize the metabolic syndrome should be in an integral fashion, including the prevention and reduction of the cardiovascular risk associated with the syndrome.



