



The consequences of pathologic adipose tissue accumulation have been described for almost all organs. Nonalcoholic fatty liver disease (NAFLD) is considered the most relevant hepatic manifestation of obesity. There is great interest in the study of NAFLD, and new insights into its pathogenic process have been described. Currently, in addition to insulin resistance, which was considered the hallmark of this disease, endocrine, immu-nologic, and central nervous system factors are attracting interest as explanatory variables. In this review, new factors associated with the main theories on the pathophysiology of NAFLD are analyzed.
The consequences of pathologic adipose tissue accumulation have been described for almost all organs, the main feature of which is a state of insulin resistance. Several consequences on hepatitis C virus infection and alcoholic liver disease for the liver, and biliary system have been described, including an increased incidence of gallstone disease. Currently, nonalcoholic fatty liver disease (NAFLD) is considered the most relevant hepatic manifestation of obesity.7 There is great interest in the study of NAFLD, and there are new insights into its pathogenic process. Currently, in addition to insulin resistance, endocrine, immunologic, and central nervous system factors are attracting interest as explanatory variables associated with this chronic liver disease. In this review, new factors associated with the main pathogenic theories on the pathophysiology of NAFLD are analyzed.
Insulin resistanceClassical clinical descriptions of the pathophysiology of NAFLD involve impaired insulin sensitivity in hepatic and muscle tissues.1-3
Numerous factors participate in the regulation of insulin sensitivity, including a complex network of endocrine signals in which adiponectin plays a key role. Structurally, adiponectin belongs to the complement 1q family4-6 and contains a carboxyl-terminal globular domain and an amino-terminal collagenous domain.7,8 The globular domain shares homology with the globular domains of collagens VIII and X. The complement 1q family characteristically forms multimers.4,5 Adiponectin regulates the activity of AMP-activated protein kinase and increases the expression of peroxisome proliferator-acti-vated receptor (PPAR)-α target genes such as CD36, acylcoenzyme A oxidase, and uncoupling protein 2 (UCP-2). Detailed descriptions of the anti-inflammatory properties of adiponectin and its effects on tumor necrosis factor (TNF)-α activity have been published.9,10 An interesting aspect of adiponectin is its ability to form multimers, which determine its physiological effect. It also undergoes extensive postranslational modification via hydroxylation and glycosylation and circulates in trim-eric, hexameric, and oligomeric forms. Mutations in the adiponectin gene, hyperglycemia, and hyperinsulinemia reduce the levels of circulating high-molecular-weight (HMW) adiponectin, the active form.11 Consequently, the plasma ratio of HMW adiponectin to total adiponectin is a stronger predictor of insulin resistance than the total concentration of adiponectin.12 HMW adiponectin concentration is inversely correlated with insulin resistance and elevation of fasting blood glucose level. Moreover, serum alanine aminotransferase (ALT) level was the strongest univariate correlate of HMW adiponectin level and was an independent covariate of both total-and HMW-adiponectin level in a multiple linear regression analysis.13 The lysyl hydroxylase family, consisting of lysyl hydroxylase-1, -2a, -2b, and -3, catalyzes the conversion of lysine into hydroxylysine and is responsible for postranslational modification of adiponectin.14 This finding is particularly interesting because there is evidence that modification of the structure of adiponectin significantly alters the phosphorylation of hepatic AMP-activated protein kinase,15 a key enzyme through which adiponectin exerts its metabolic effects.
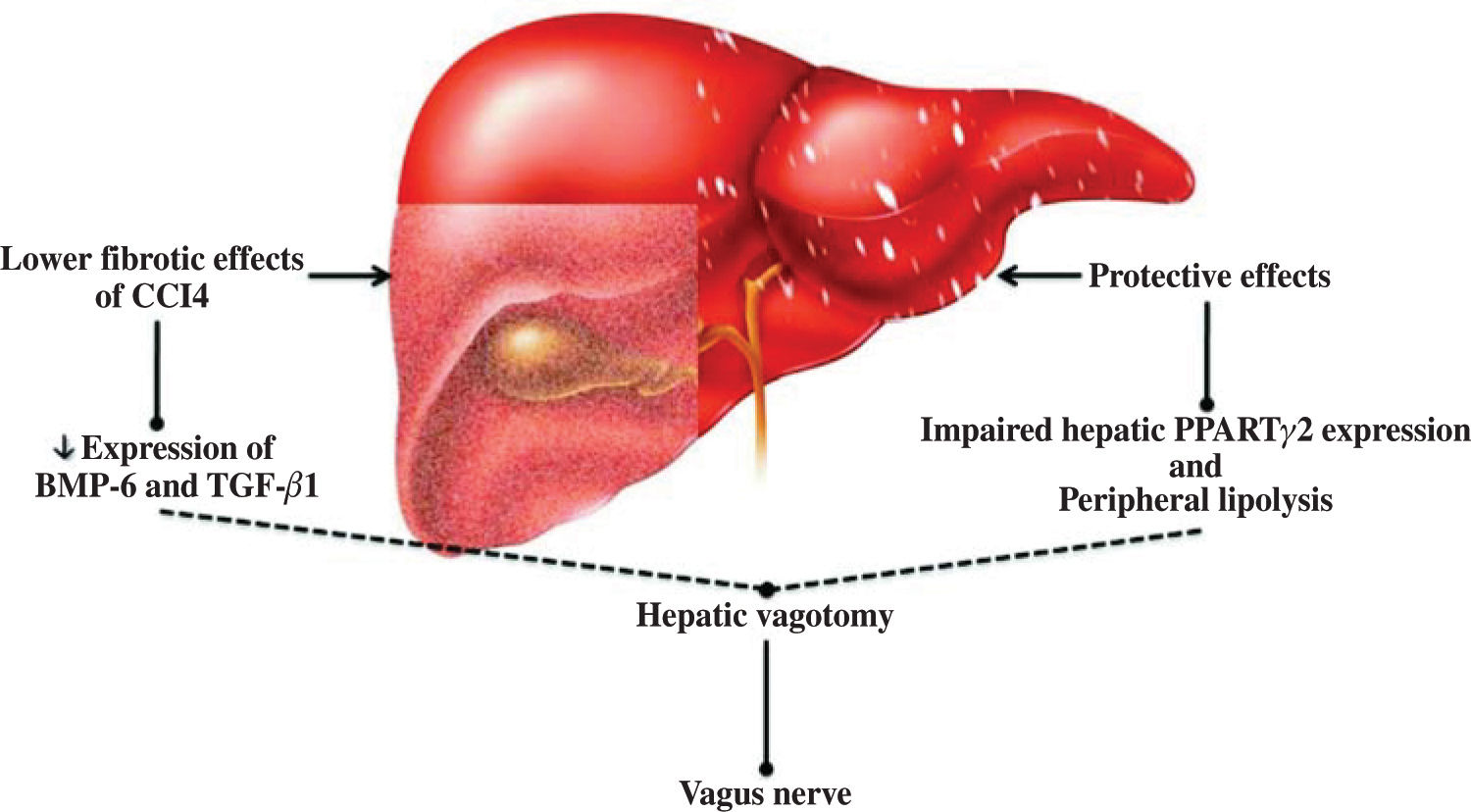
The discovery of leptin has made an important contribution to our understanding of the relationship between the nervous system and obesity, and therefore of the relationship between the nervous system and NAFLD.16,17 In this respect, the study of Uno et al.18 demonstrated the direct role of the vagal afferent and efferent neural branches in the regulation of liver insulin sensitivity. They performed selective hepatic vagotomy on mice in which PPARγ2 was overexpressed using an adenovirus vector and administered a pharmacologic β-adrenergic blocker. They demonstrated that 1) β-adrenergic nerve function enhances lipolysis in adipose tissues of mice that overexpress PPARγ2, 2) the hepatic vagus nerve mediates the remote effects of hepatic PPARγ2 expression, 3) activation of the afferent vagal nerve at the liver mediates the remote effects of hepatic PPARγ2 expression on peripheral lipolysis, and 4) the effects of PPARγ2 on glucose metabolism are dependent on the afferent vagal and efferent sympathetic nerves. This hypothesis was extended not only to the insulin sensitivity pathway (Figure 1) but also to liver fibrosis, as confirmed by other studies in which both surgical and chemical methods of cholin-ergic denervation decreased expression of bone morpho-genetic protein-6 and transforming growth factor-/31 in carbon tetrachloride-induced liver fibrosis of rats.19 This mode of action has also been described for other elements of metabolic syndrome.20,21
 and transforming growth factor (TGF-β1) expression. In nonalcoholic fatty liver disease, this protective effect may involve peroxisome proliferator-activated receptors (PPAR) and peripheral lipolysis.")
Regulation of liver physiology by the vagus nerve. Experimental evidence suggests that the vagus nerve plays a protective role against adipose tissue accumulation and chemical liver damage. This effect is mediated by a limited fibrosis response and downregulated bone morphogenetic protein-6 (BMP-6) and transforming growth factor (TGF-β1) expression. In nonalcoholic fatty liver disease, this protective effect may involve peroxisome proliferator-activated receptors (PPAR) and peripheral lipolysis.
Despite the relationship between insulin resistance and overaccumulation of fatty acids in the liver, the mechanisms involved in this phenomenon have not been completely described. Hyperinsulinemia caused by dietary overindulgence results in 1) increased microsomal triglyceride transfer protein (MTP) expression, 2) increased apoB availability, and 3) increased triglyceride levels caused by transcriptional induction of lipogenic enzymes. Most of the flux of fatty acids originates from adipose tissue under normal circumstances. However, during insulin resistance, de novo lipogenesis and lipo-protein uptake make significant contributions to the fatty acid pool, resulting in steatosis. During the insulin resistant state, the liver adapts by increasing MTP expression via nuclear localization of the forkhead box-containing protein O subfamily-1 (FoxO1) and inducing MTP transcription.22 This is concordant with the results of studies of humans, which show that FoxO1 expression differs according to liver histopathology: the level of FoxO1 is greater in patients with nonalcoholic steatohepatitis (NASH) than in NAFLD patients or subjects who do not have NAFLS. This may be related to increased levels of transcription NAFLD factors such as sirtuin-1. Moreover, FoxO1 mRNA levels are correlated with insulin resistance markers, NASH activity score, and the percentage of stea-totic hepatocytes but not with the severity of fibrosis.23
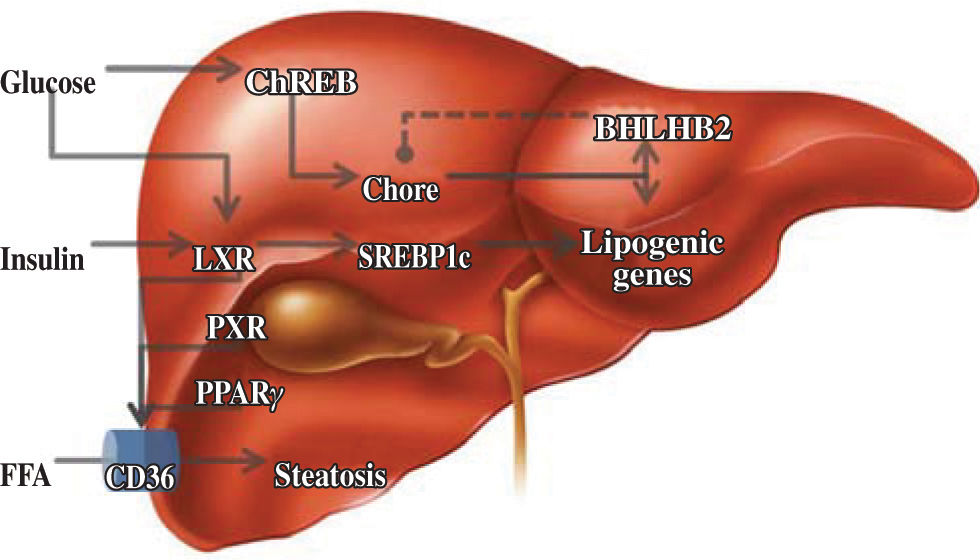
Lipid metabolismThe liver is responsible for the conversion of excess dietary carbohydrates to triglycerides (TGs) through de novo lipogenesis, a mechanism that is regulated by glucose and insulin.24 In this hormone-controlled pathway, the nuclear receptors are ligand-activated transcription factors that coordinate gene expression in response to hormonal and environmental signals. Members of the su-perfamily that forms heterodimers with the retinoid X receptor (RXR) serve as sensors of dietary components.25 However, this pathway was mainly characterized on the basis of the effects of insulin on the enzymatic pathway involving the sterol regulatory element binding protein-1c, which only partially explains fatty acid synthesis.26 Although the nature of the glucose-signaling compound is not known, the identification of a glucose-responsive basic/helix-loop-helix/leucine zipper transcription factor named ChREBP (carbohydrate responsive element binding protein) has shed light on the mechanism whereby glucose affects gene transcription. ChREBP is a large protein (864 amino acids) that contains several domains, including a nuclear localization signal near the N-termi-nus, polyproline domains, a basic loop-helix-leucine zipper, and a leucine-zipper-like domain. Glucose activates ChREBP by regulating its entry from the cytosol into the nucleus, thereby promoting its binding to the carbohydrate responsive element in the promoter regions of glyc-olytic and lipogenic genes.27 Glucose regulatory mechanisms also involve the liver X receptor (LXR), a nuclear receptor that coordinates hepatic lipid metabolism. Mitro et al.28 studied HepG2 cells transfected with expression vectors for LXR and RXR, and demonstrated that glucose and its derivatives induce LXR and RXR activity in cells grown in the absence of glucose. They also demonstrated that this is mainly a direct effect of glucose and its derivatives on LXR activity and not a result of generation of an endogenous ligand or postranscriptional modification. It is interesting to note that LXR binds to more than one site and may act in combination with another ligand. This effect on lipogenesis is mediated by increased expression of ChREBP, indicating that the concerted action of sterol regulatory element binding pro-tein-1c and ChREBP is necessary for normal fatty acid and triglyceride synthesis in vivo. Subsequent evidence has clarified some aspects of the regulatory effect of glucose on hepatic lipid metabolism. Using LXRα/β knockout mice, Denechaud et al.29 demonstrated high carbohydrate diet-mediated induction of ChREBP expression and translocation. Interestingly, when ChREBP was silenced by siRNA, lipogenic genes were not induced by high glucose concentrations in LXRα/β knockout hepatocytes, confirming the importance of ChREBP as a glucose sensor. This demonstrates the lack of involvement of LXR in the regulation of glucose-sensitive genes in the liver. A new element involved in this regulatory pathway is the transcription repressor, basic helix-loop-helix binding Protein 2, mRNA levels of which are elevated in the muscles of diabetic and insulin-resistant humans.30 Iizuka et al.31 showed that overexpression of basic helix-loop-helix binding Protein 2 in rat hepatocytes inhibits the expression of glucose-induced lipogenic genes by inhibiting the binding of ChREBP to the carbohydrate responsive element. Basic helix-loop-helix binding Protein 2 competes with ChREBP for binding to the carbohydrate-responsive element and suppresses the activities of fatty acid synthase and hepatic pyruvate kinase. This suggests that there is a negative feedback loop between ChREBP and basic helix-loop-helix binding Protein 2, but its significance is unclear at present (Figure 2).
, and this pathway is apparently regulated by negative feedback by basic helix-loop-helix binding Protein 2 (BHL-HB2). Other activators of nuclear receptors are insulin and, indirectly, the fatty acids, which increase expression of the membrane transporter, CD36.")
Regulatory elements of lipid metabolism. Glucose has important effects on carbohydrate responsive element binding protein (ChREB), and this pathway is apparently regulated by negative feedback by basic helix-loop-helix binding Protein 2 (BHL-HB2). Other activators of nuclear receptors are insulin and, indirectly, the fatty acids, which increase expression of the membrane transporter, CD36.
Small heterodimer partner (SHP) is an unusual orphan nuclear receptor in that it lacks a DNA-binding domain. It is expressed in several tissues and has a relatively high level of expression in the liver, heart, and pancreas. SHP interacts with a number of other nuclear receptors, including estrogen receptor α, RXRα, hepatocyte nuclear factor-4α, liver receptor homolog-1, and estrogen receptor-related receptor γ, and inhibits their transcriptional activity. Nuclear receptors are key regulators of metabolic pathways. Studies with SHP knockout mice have confirmed that SHP is involved in the homeostasis of bile acid, cholesterol, and TG. In humans, loss of SHP function has been associated with mild obesity in Japanese subjects, but genetic variation in SHP activity is not commonly associated with obesity in Caucasians in the United Kingdom. SHP inhibits the function of a variety of nuclear receptors that act as metabolic regulators. Previous studies of SHP-/- mice have identified functions for SHP in the negative regulation of bile acid production, lipo-genesis, and energy production. A recent study involving a hepatic SHP overexpression model suggests that SHP plays an important role in the control of TG metabolism.32 Hartman et al.33 studied the role of SHP in liver lipid metabolism using SHP, farnesoid X receptor (FXR), and low density lipoprotein (LDL) receptor knockout mice. In this study, levels of markers of hepatic inflammation, including serum ALT and aspartate aminotrans-ferase (AST), were elevated in the FXR-/- mice but not in the SHP-/- mice, suggesting that the loss of SHP had an offsetting beneficial activity on plasma lipids and hepatic inflammation. When fed a challenge diet containing cholic acid, the expression of several inflammatory marker genes, including vascular cell adhesion molecule, intracellular adhesion molecule-1, and TNFa, was increased in the livers of the SHP+/+ mice. The basal level of expression of these genes was not altered in the SHP/- mice, and there was no induction of these genes in the SHP-/- mice by the diet containing cholic acid. In the SHP+/- mice, the diet containing cholic acid induced vascular cell adhesion molecule expression to the same magnitude as in the SHP+/+ mice. In contrast, induction of intracellular adhesion molecule-1 and TNFα was partially reduced in the SHP+/- mice. These results suggest that various inflammatory genes may differ in sensitivity to the loss of SHP. This thesis was also confirmed with a Western diet, which also increased hepatic expression of hepatic inflammatory marker genes, including vascular cell adhesion molecule, intracellular adhesion molecule-1, and TNFα in the LDL receptor-/- mice. Again, the loss of SHP expression in the LDLR/SHP-/- mice resulted in a near complete block in the induction of these genes. This protective effect was confirmed in another animal model, the leptin-deficient model, in which it was observed that loss of SHP reduces brown fat mass without affecting obesity. Leptin-deficient animals that lacked SHP also had improved insulin sensitivity. Another interesting finding is the effect of SHP on MTP; basal MTP protein levels were increased in both SHP-/- and OB-/-/SHP-/- livers compared with SHP +/+ or OB-/- controls. By introducing mutations of the liver receptor homolog-1, the authors demonstrated that this receptor binds to the MTP promoter and increases MTP expression in hepatocytes and that this transactivation is repressed by SHP.
As previously mentioned, activation of orphan nuclear receptors, including LXR, PPAR, and the pregnane X receptor (PXR), has been associated with hepatic steato-sis. PPARγ2 induces lipid accumulation in hepatocytes. One of the lipogenic genes induced by PPAR is Cd36, a membrane receptor capable of taking modified forms of LDL and fatty acids up from the circulation. Moreover, LXR has been reported to activate PPARγ2 in the liver. Using LXR transgenic, PXR transgenic, and Cd36 null mice, Zhou et al.34 investigated the role of CD36 as a key step in the regulation of LXR, PXR, and PPARγ in hepatic steatosis. They observed that in mice transfected with LXR and wild-type mice treated with an LXR agonist, there was a significantly increased expression of CD36 (a phenomenon conserved in primary human hepato-cytes), and this was related to the activation of endogenous CD36 by the LXR agonist in primary human hepatocytes (Figure 2). To demonstrate that CD36 is an essential factor in the promotion of steatosis via LXR, Cd36 null and wild-type mice were treated with an LXR agonist. Cd36 null mice had greatly attenuated LXR-agonist-induced hepatic lipid accumulation, which is concordant with the observation that CD36 null mice failed to accumulate in-trahepatic TG.
Finally, a new element connecting hepatic lipid metabolism, xenobiotic metabolism, and fatty liver diseases is described. To protect the body against xenobiotics and the buildup of toxic endogenous lipids, two nuclear receptors function in a metabolic cascade to regulate detoxification and elimination. The constitutive an-drostane receptor (CAR) mediates the response to a narrow range of phenobarbital-like inducers. Although CAR was initially proposed to be constitutively active, ligands with negative (androstanes) and positive (phenobarbital) effects are present. CAR binds to and activates the CYP2B promoter in response to phenobarbit-al-like molecules, the pesticide 1,4-bis[2-(3,5-dichlo-rpyridyloxyl)]- benzene (TCPOBOP), certain androgens, and the muscle relaxant drug, zoxazolamine. Genetic disruption of the mouse CAR gene abolishes induced CYP2B expression, resulting in increased serum levels of nonmetabolized products,35 and also acts as a het-erodimer with the RXR. This suggests that this nuclear receptor plays a critical role in the pathogenesis of NASH because of its ability to regulate genes. Yamaza-ki et al.36 studied CAR -/- mice fed a methionine-and choline-deficient diet (MCDD), and observed higher levels of ALT in CAR+/+ mice and CAR+/+ mice that had been treated with an agonist of CAR than in CAR-/- mice. Consistent with serum ALT levels, inflammatory cell infiltration and hepatocyte necrosis in the CAR+/+ mice was more severe than in the CAR-/- mice. The inflammation score for the CAR -/- mice was lower than that for the CAR+/+ mice. Both the CAR+/+ and CAR-/- mice fed the MCDD had marked steatosis compared with the mice fed the control diet, and this effect was independent of the hepatocyte fat overload. Thus, this is a good model of the second step in the two hits theory. All these deleterious effects are associated with an elevation in lipid peroxidation and the induction of cyto-chrome P450 and inducible nitric oxide synthase.
These results support the importance of lipid metabolism in NASH. However, in studies of disturbances in intestinal tissue using mice fed a high-fat diet (HFD), microarray analysis demonstrated differential gene expression in the middle part of the small intestine during dietary intervention. Dietary fat had the strongest effect in the proximal and middle part of the small intestine. In these parts, genes involved in fatty acid transport, chylo-micron synthesis, and especially fatty acid oxidation were highly upregulated, whereas genes involved in cholesterol transport were downregulated by dietary fat. Vil-lus length and the total number of cells per villus were significantly higher in mice fed the HFD, which is in concordance with the increased cell proliferation rate observed in HFD mice. Together, these data indicate that fat-induced inhibition of apoptosis and increased cell proliferation results in enlargement of small intestinal villi. This may increase the capacity for fat absorption.37
Immune responseThe role of the immune response in chronic liver diseases, including NAFLD, has been studied in several animal-based experiments and human trials, which have demonstrated the importance of several cytokines in the inflammatory component of NAFLD. However, the regulatory process associated with this immune-mediated damage is not completely understood. New information on the roles of the signal transducers and activators of transcription (Stats) is important for understanding the relationship between obesity and liver diseases. There are seven Stat transcription factors (Stat1, Stat2, Stat3, Stat4, Stat5a, Stat5b, and Stat6), all with six shared domains of homology. This characteristic differs from that of the four Janus tyrosine kinases (Jaks) (Jak1, Jak2, Jak3, and Tyk2), which have seven Jak homological (JH) domains and play a role in receptor binding activity. Binding of cytokines to cytokine receptors attracts and activates one or more of the Jaks, which are tyrosine phosphorylated themselves, and phosphorylate tyrosines on the cytokine receptor sites, thereby creating active docking sites for Stats.38 This pathway is apparently associated with the effects of leptin. The involvement of leptin in NAFLD has been clearly described; the main profile of NAFLD patients is hyperleptinemia with resistance to the physiologic effects of leptin,17·39·40 but this resistance profile has not been clearly described. Using rats fed a fructose- vs. glucose-supplemented diet, Roglans et al.41 showed that fructose-fed rats were hypertriglyceridemic and hyperlep-tinemic and had TG accumulation in liver tissue. This was associated with a marked reduction in the activity of the hepatic β-fatty acid oxidation system because of decreased PPAR-α transcriptional activity, and consequently decreased activation of nuclear factor (NF)-κB. Fructose-fed rats showed impairment of the leptin transduction signal through the STAT3 pathway. Leptin administration to rodents has been shown to increase liver fatty acid oxidation and to decrease hepatic steatosis by activating the PPAR-α system, and to diminish the expression of several lipogenic genes. This confirms the hypothesis that the activation of STAT3 transcriptional activity is necessary for leptin’s effects on energy homeo-stasis,42 and this activity may be involved in the action of other cytokines such as interleukin 6.43 These data indicate a clear role for leptin in STAT3 activation, particularly as data from leptin-deficient animal models show that STAT3 is overexpressed in these animals.44 Moreover, these STAT signaling effects are observed not only in the liver but also in the central nervous system, as basal hypothalamic phosphorylated STAT3 levels are significantly higher in high-fructose-fed animals than in controls, whereas leptin receptor protein levels were similar between the control and fructose groups, which affected weight gain, adiposity, and anorexia.45 This information has now been applied to clinical research. Gene variants and haplotypes involved in linkage-disequilibrium blockade of STAT3 have been shown to be involved in NAFLD. The markers, rs6503695 and rs9891119, have been shown to be associated with NAFLD in that rs6503695-T and rs9891119-A allele carriers are 2.3-and 2.5-fold, respectively, more likely to have NAFLD than noncarriers. Additionally, these gene variants are associated with the clinical and histological features of NAFLD.46
The importance of organs other than adipose tissue in NASH, particularly the intestine and the immune system, has been described recently. As blood leaving the gut empties directly into the portal vein, the liver is exposed to gut-derived endotoxin. As a result of endotoxemia, Kupffer cells are activated via the Toll-like receptor (TLR) 4 complex on the cell surface. This receptor is a member of the Toll-like family of pattern recognition receptors that are of central importance during host defense against invading pathogens. TLR-4 interaction with en-dotoxin results in the release of a myriad of proinflamma-tory mediators that induce hepatic injury and fibrosis. In addition, cytokines have profound effects on lipid metabolism. This hypothesis has been confirmed using mice fed the MCDD, as plasma endotoxin levels in these mice are threefold greater than those of control diet mice. Additionally, TLR-4 expression was increased five-fold by MCDD with similar changes at protein level. The expression of the TLR-4 accessory molecules, MD-2 and CD14, was also increased after feeding the MCDD. C3H/HeJ mice, which lack TLR-4 signaling because of a spontaneous point mutation, were used to confirm the relevance of TLRs in hepatic damage in a NASH model. In wildtype mice, the MCDD resulted in extensive macrovesicu-lar steatosis and necrosis typical of steatohepatitis. These histopathological changes were largely absent in the C3H/HeJ mice. Feeding the MCDD resulted in clustering of neutrophils at sites of steatosis and injury in wild-type mice. Macrophage accumulation in the close vicinity of injured hepatocytes was distinctly more pronounced. The infiltration of both neutrophils and macrophages was blunted in C3H/HeJ mice fed the MCDD; apparently PPAR-α may be negatively regulated in response to TLR-4 signaling.47 As previously discussed, Kupffer cells are important elements in the development of immune response in NASH, an effect that may be TNFa dependent. In mice deficient in both TNFα-receptors (TNFR1 and TNFR2) (TNFRDKO mice), the administration of the MCDD resulted in a significant increase in liver TG levels in both wild and TNFRDKO mice compared with levels observed after administering the control diet. The livers of wild-type mice exhibited increased collagen deposition localized around the central vein and throughout the lobule in a pericellular distribution.
In contrast, the extent of centrizonal fibrosis was markedly reduced in livers of TNFRDKO mice. The livers of wild-type mice fed the MCDD also exhibited increased staining for a-smooth muscle actin, TNFR1 and TNFR2 deficiency, but reduced fibrotic damage. The administration of the MCDD augmented the number of large macrophage clusters preferentially distributed in pericentral regions, and TNFR1/TNFR2 deficiency reduced this recruitment. The reduction in recruitment may be explained by increased hepatic CD14 expression (the number of CD14, a functional lipopolysaccharide receptor, is enhanced on Kupffer cells after activation by multiple stimuli such as lipopolysaccharide and TNFa) and lower expression of hepatic intracellular adhesion molecule 1 and vascular cell adhesion molecule 1 in knockout mice.48
Another pathway associated with the immune response in NASH that has not been investigated in detail involves Chitotriosidase (Chit), an enzyme that belongs to the family of glycosylhydrolases. The CHIT gene is localized on chromosome 1q31-q32,10 and it consists of 12 exons and spans about 20 kb of genom-ic DNA. Tissue macrophages largely secrete newly synthesized 50 kDa Chit, and a second step cleaves the enzyme to produce the active form of Chit (39 kDa). In several diseases, pathological tissue macrophages express Chit on a massive scale. In humans, levels of Chit expression are significantly higher in NASH patients than NAFLD patients or control patients, and there is a positive correlation between CHIT expression and the degree of NASH. Plasma levels of Chit activity are higher in NASH patients than in NAFLD patients. The expression of TNFa correlates with Chit induction in NASH and steatosis patients, whereas in control subjects, there is no correlation between CHIT and TNFa mRNA levels.49
Oxidative stressBecause of the key role that mitochondria play in fatty acid metabolism, these structures are involved in the pathogenesis of NAFLD. When animals are fed a high-fat diet, the state 3 respiratory capacity and respiratory control ratio values increase. The respiratory quotient and the nonprotein respiratory quotient are altered, indicating that there is a shift in oxidation of nutrients. However, these changes appear to be affected by circadian rhythm and age in rats.50 The role of oxidative stress and mitochondrial dysfunction has been described widely, but not all processes involved have been clarified, particularly the role of oxidative stress in diseases associated with obesity. In mice fed an HFD, increased production of reactive oxygen species (ROS) precedes the elevation of TNFa and free fatty acids in the plasma and liver. Gene expression levels of various metabolic pathways increase after 6 weeks on the HFD.
In particular, pathways for sterol regulatory element binding protein 1c-related fatty acid synthesis and PPA-Rα are upregulated in the livers of mice fed an HFD. In contrast, the pathway for fatty acid synthesis is down-regulated in adipose tissue. In contrast to the pathways involving fatty acid metabolism, oxidative stress pathways are coordinately upregulated in both the liver and adipose tissue, but the mRNA expression level of TNFα in the liver and adipose tissue of mice fed an HFD for 6 weeks is similar to that of control mice. Furthermore, the plasma level of TNFα was below the detection limit of the enzyme-linked immunosorbent assay in both groups. These data suggest that the production of ROS may be an initial key event that triggers HFD-induced insulin resistance.51 These modifications are accompanied by functional changes. In animals fed an MCDD, the state 3 respiratory rate after the addition of ADP was increased by 45% by either glutamate/malate or succi-nate/malate. Either of the following two mechanisms may be involved in this decrease in mitochondrial efficiency: a change in the proton conductance of the inner mitochondrial membrane, i.e., a proton leak, or a decrease in the efficiency of coupling between electron and proton flux through the respiratory chain, i.e., redox slipping.52
Two other new elements involved in this complex process have recently been described. Degradation of serotonin is catalyzed by the mitochondrial enzyme, monoamine oxidase A (MAO-A), generating 5-hydroxy-indolic acid and ROS such as hydrogen peroxide. ROS generated by MAO-A-mediated catabolism of serotonin were recently reported to play a pivotal role in cardiomy-ocyte death; this has also been observed in hepatocytes. With tryptophan hydroxylase 1 (Tph1) knockout mice (Tph1-/-), i.e., serotonin-deficient mice, are fed a choline-and methionine-deficient diet, they develop steatohepa-titis and upregulation of MAO-A transcripts and protein level. This is associated with lower malondialdehyde levels in CMD-fed Tph1-/- animals. Taken together, these data strongly suggest that increased hepatic uptake and catabolism of serotonin is an important source of oxida-tive stress in this model of steatohepatitis. Another important element is UCP2, an inner mitochondrial membrane protein with ubiquitous tissue distribution, which mediates proton leakage across the inner membrane by uncoupling substrate oxidation from ATP synthesis. When a choline-deficient diet was administered, UCP2 was expressed at a low level in normal hepatocytes but was upregulated at weeks 7 and 11 in NASH. Hepatic ATP content was consistently lower in NASH rats than in control rats at 3, 7, and 11 weeks. Interestingly, ATPase activity was unaffected at weeks 3 and 7 on NASH compared with the controls, and then it increased relative to the controls at week 11. This pattern may be explained by an interaction with 4-hydroxy-2-nonenal-protein ad-ducts. Overexpression of UCP2 in hepatocytes during the rogression of NASH causes a proton leak to avoid a progressive increase in the rate of mitochondrial H2O2 production.53
ApoptosisApoptosis is a mode of cell death used by multicellu-lar organisms to dispose of unwanted cells under a diversity of conditions, including NAFLD. In subjects with NASH, the number of TUNEL-positive cells is greater than that of controls.54 The importance of this process is illustrated by clinical trials that have shown that the substrates of proteases, particularly cytokeratin-18 (CK-18), are very good markers of NASH. In patients with biopsy-proven NASH, plasma levels of CK-18 fragments ranged from 105.5 to 2,306.4 U/L. These levels were significantly higher than those observed in 35 healthy controls (median 234 U/L).55 Similar results have been reported for tissue polypeptide-specific antigen (TPS), a serological mirror of keratin18, and it has been shown that its accuracy in differentiating between NASH and simple fatty liver is better than any other parameter. A TPS value of 88 ng/mL among patients with underlying NAFLD was associated with a high probability of NASH (sensitivity and specificity, 92 and 96%, respectively).56
This clinical evidence indicates that this pathway is relevant to the pathophysiology of NASH. Apoptosis is associated with oxidative stress. In liver cells cultured in enriched medium with free fatty acids, the unfolded protein response (UPR) is initiated by three endoplasmic reticulum (ER) transmembrane proteins, inositol-requir-ing ER-to-nucleus signaling protein 1a (IRE1a), RNA-de-pendent protein kinase-like ER eukaryotic initiation fac-tor-2a kinase (PERK), and activating transcription factor-6 (ATF6). Activation of IRE1 promotes the splicing of X-box-binding protein-1 (XBP1) mRNA and subsequent transcription of molecular chaperones and genes involved in ER-associated degradation. Increased expression of GADD34, a member of the growth arrest and DNA damage family of proteins, is involved in the dephos-phorylation of eIF2a, and therefore in reversal of transla-tional attenuation. Failure of the UPR to reestablish ER homeostasis may result in apoptosis via mitochondrial-independent and-dependent mechanisms.57 The precise mechanism of apoptosis in NASH has been partially described. Overexpression and subsequent translocation of the proapoptotic protein Bax induces cytochrome c release from mitochondria and, consequently, caspase-3 activation. Caspase-3 activation coincides with a 2.3-fold upregulation of Bax in NASH patients. In hepatocytes, apoptosis is usually initiated by interactions between plasma membrane receptors such as TNF-R1 and Fas and their ligands, TNFa and the Fas ligand, respectively. These signals are combined in a final common apoptotic pathway that targets mitochondria, promoting cyto-chrome c release and activation of a caspase cascade.
Liver injury is often linked with the induction of protective factors such as antiapoptotic members of the Bcl-2 protein-family. Bcl-2 is notably upregulated in NASH (an increase of 112-fold) and is almost absent in control specimens. Despite upregulation of Bcl-2, apoptosis is increased in steatohepatitis, which confirms that the protective response is insufficient under these conditions. In contrast to Bcl-2, no significant changes were found in Bcl-xL expression, suggesting that this protein is not implicated in a possible protective liver pathway against steatohepatitis.54 New advances have been made since the initial description of the role of Fas in NASH, in which upregulation of Fas contributes to hepatocyte apoptosis. Zou et al.58 analyzed pathways involved in Fas-related damage. They detected the Met-Fas complex in the majority of normal human samples but not in simple stea-totic liver samples. Fas was also dissociated from Met in almost all NASH cases. Fas abundance was similar in control and diseased livers, whereas Met was significantly less abundant in fatty livers and NASH livers than in normal control livers. The dissociation of Met from Fas showed that Fas and Met were colocalized in normal but not in fatty hepatocytes. Hepatocyte growth factor (HGF) expression was greater in fatty liver tissue than in normal liver tissue, suggesting that the vulnerability of fatty hepatocytes to apoptosis is not caused by a lack of Met activation by its ligand, HGF. To confirm that fatty acid administration induces dissociation of Met from Fas and the marked stimulation of FasL expression, they used mutant and functional peptides from Met YLGA, a pep-tide that binds specifically to Fas. The functional peptide reduced the apoptotic population induced by FasL from 31.12 to 6.17%, whereas the mutant peptide had no effect. When this functional peptide was administered to ob/ob mice, the activation of caspase-8 and caspase-3 was significantly reduced, and this was accompanied by a significant reduction in hepatocyte apoptosis and liver damage (reduced hepatic inflammation and serum ALT level), lower hepatic caspase-3 activity, and a lower number of TUNEL-positive hepatocytes. These changes were associated with a 300% reduction in collagen-1 mRNA expression.



