





La muerte súbita (MS) es un evento trágico que representa un grave problema de salud. Se estima que causa cerca de 4-5 millones de decesos por año en todo el mundo.
La MS se define como la muerte ocurrida en el lapso de 1h en una persona sin signos previos de fatalidad; puede denominarse «recuperada», cuando el paciente afectado sobrevive al episodio potencialmente fatal ya sea por reanimación cardiopulmonar o desfibrilación efectiva.
Las canalopatías arritmogénicas son alteraciones funcionales de los canales iónicos del corazón, generalmente condicionados por mutaciones en los genes que los codifican y dan lugar a diversos tipos de arritmias que pueden culminar en MS, el deceso ocurre normalmente antes de los 40 años y el corazón en estudio de autopsia suele ser estructuralmente normal. En la presente revisión presentamos las principales causas de MS en el contexto del corazón estructuralmente normal y discutimos el abordaje que se debe dar a los pacientes y familiares de víctimas que han experimentado éste trágico evento.
Sudden death (SD) is a tragic event and a world-wide health problem. Every year, near 4-5 million people experience SD.
SD is defined as the death occurred in 1h after the onset of symptoms in a person without previous signs of fatality. It can be named «recovered SD» when the case received medical attention, cardiac reanimation effective defibrillation or both, surviving the fatal arrhythmia.
Cardiac channelopathies are a group of diseases characterized by abnormal ion channel function due to genetic mutations in ion channel genes, providing increased susceptibility to develop cardiac arrhythmias and SD. Usually the death occurs before 40 years of age and in the autopsy the heart is normal. In this review we discuss the main cardiac channelopathies involved in sudden cardiac death along with current management of cases and family members that have experienced such tragic event.
La muerte súbita (MS) es un evento trágico que representa un grave problema de salud mundial1. En las últimas décadas se ha logrado un avance importante en el entendimiento de las bases genéticas y fisiopatológicas de este problema que, aunado a un claro avance tecnológico, ha tenido una repercusión clave en el tratamiento y en la prevención tanto primaria como secundaria de la MS. A pesar de esto, se estima que la MS causa cerca de 4-5 millones de decesos por año en todo el mundo2.
Los atletas de alto rendimiento representan un grupo en particular con mayor vulnerabilidad a la MS, hecho reconocido por los antiguos griegos quienes documentaron la MS de Pheidippides tras haber corrido una distancia cercana a los 40km desde la ciudad de Maratón hasta Atenas para reportar la victoria de Grecia sobre los persas (fig. 1).

En la presente revisión presentamos los principales mecanismos involucrados en la MS cardiaca (MSC) y discutimos el abordaje contemporáneo de los pacientes y familiares de víctimas que han experimentado este trágico evento. Hemos realizado una búsqueda bibliográfica sistemática en PubMed de artículos originales, revisiones y guías sobre las bases genético-moleculares y el manejo actual de la MS para ofrecer al lector un panorama general del problema.
Definición de muerte súbita cardiacaLa definición más aceptada de MSC la define como la que ocurre en el lapso de 1h en una persona sin signos previos de fatalidad3. El término se refiere a la súbita interrupción de la actividad cardiaca; la persona deja de responder a estímulos, sin esfuerzo respiratorio y sin signos circulatorios4. La MSC típicamente se presenta en individuos con un sustrato anatómico o electrofisiológico subyacente, sin un evento desencadenante identificable1.
La fibrilación ventricular es la consecuencia final. La supervivencia disminuye un 10% por cada minuto que el paciente permanece con esta arritmia5. Diferentes taquicardias, bradicardias y actividad eléctrica sin pulso también pueden asociarse con la interrupción de la actividad mecánica cardiaca en ausencia de signos circulatorios. En ocasiones puede ser difícil de distinguir la MSC de otras causas de MS no cardiaca que incluyen eventos vasculares cerebrales, tromboembolia pulmonar y rotura aórtica entre otras.
La MSC puede denominarse «recuperada» cuando el paciente afectado ha recibido atención médica oportuna, con reanimación cardiopulmonar o desfibrilación efectiva y sobrevive al evento fatal.
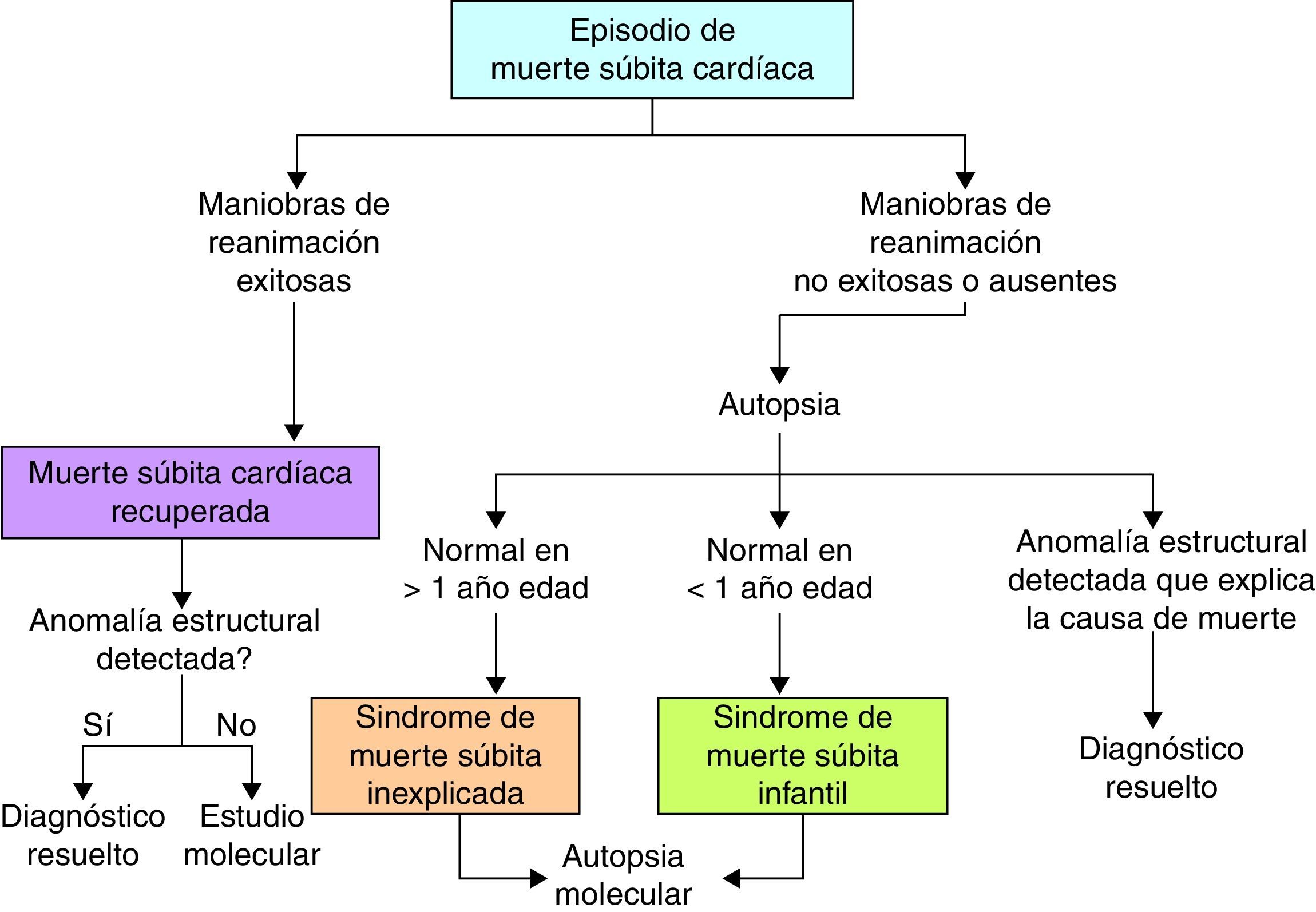
Cuando se ha realizado una autopsia completa, la MSC tiene otra connotación y es importante aquí incluir otros 2 términos (fig. 2): El síndrome de la MS inexplicada que se refiere a los pacientes mayores de un año de edad con MS en los que, a pesar de haberse practicado estudio de autopsia, la causa de muerte aún no puede definirse; y el síndrome de la MS infantil, que se refiere a los pacientes menores de un año con MS, en quienes la causa no es identificada a pesar del estudio de la autopsia. En este contexto, la muerte puede aun deberse a causas cardiacas no identificables macroscópicamente, como revisaremos más adelante.

En cuanto al género, en términos generales es más frecuente en varones (60%) que en las mujeres (40%), pero esta predominancia varía en magnitud dependiendo de la edad6.
Enfermedades estructurales a descartar en la muerte súbitaEn general el hallazgo clínico más comúnmente asociado a la MS es la enfermedad arterial coronaria, presentándose en aproximadamente el 80% de las muertes. Otro 10-15% ocurre en pacientes con cardiomiopatías, tales como miocardiopatía hipertrófica, cardiomiopatía dilatada, displasia arritmogénica del ventrículo derecho y las enfermedades miocárdicas infiltrativas (sarcoidosis, amiloidosis). El restante 5-10% lo constituyen algunas anormalidades cardiacas congénitas y principalmente las canalopatías cardiacas que por lo general cursan con un corazón estructuralmente normal2,6.
La estratificación por edad es importante pues puede orientar a la causa que ha condicionado la muerte: la causa más común en mayores de 40 años es la cardiopatía isquémica, seguida de cardiomiopatías; mientras en menores de 40 años la cardiopatía isquemia es rara, las miocardiopatías son predominantes y existe mayor prevalencia de canalopatías que constituyen el grupo de enfermedades que condicionan un corazón estructuralmente normal pero susceptible a arritmias letales motivo de esta revisión.
Síndrome de muerte súbita inexplicadaGeneralmente ocurre en pacientes menores de 40 años, tiene una devastadora repercusión en las familias, personal de la salud y comunidad en general, y suele atraer la atención del público y medios de comunicación7,8. Casi siempre ocurre en personas aparentemente sanas sin patología previa detectada; llegar al diagnóstico específico es particularmente desafiante ya que una gran proporción de pacientes son por lo demás asintomáticos y muchos pacientes no tienen evaluaciones cardiológicas apropiadas previas. Cerca de un 30% de los pacientes con MS inexplicada tienen mutaciones en canales iónicos9, por lo que en estos pacientes es importante realizar estudio genético10 o autopsia molecular (fig. 2), en busca de canalopatías arritmogénicas con el fin de prevenir potenciales MS en otros miembros de la familia. Otro abordaje de utilidad es la evaluación directa de los familiares cercanos en busca de datos clínicos sugestivos de canalopatías arritmogénicas11. Sin embargo, de acuerdo con varios estudios como el CASPeR, la investigación clínica es más efectiva que el cribado genético para orientar al clínico hacia el diagnóstico correcto12, incluso el estudio genético puede optimizarse cuando se tiene una sospecha diagnóstica, por lo que se aconseja que las pruebas genéticas se realicen solo después del estudio clínico11.
Síndrome de muerte súbita infantilEs una de las principales causas de muerte en el periodo posnatal en países desarrollados. Se define como la ocurrida en menores de un año, con un pico de incidencia en los primeros 3 meses de edad. Se ha descrito que en su presentación existen 3 factores importantes: un infante susceptible, expuesto a ciertos factores exógenos, en un periodo crítico del desarrollo, son los factores que generan las condiciones para este fatal desenlace13. Existen múltiples causas conocidas asociadas a este síndrome, y el riesgo se incrementa cuando hay un bajo peso al nacer, exposición al humo del tabaco, posición prona al dormir y es más común en la raza negra. Dentro de las causas relacionadas con la susceptibilidad del infante se han estudiado trastornos en los canales iónicos. En general se considera que cerca de un 10% de los pacientes con síndrome de la MS infantil se deben a canalopatías arritmogénicas14,15; las mutaciones en el canal de sodio predominan dentro de las anomalías de los canales iónicos16.
Principales canalopatías arritmogénicas asociadas a muerte súbitaLas canalopatías arritmogénicas son alteraciones de la función de los canales iónicos del corazón, generalmente condicionados por mutaciones en los genes que los codifican y que condicionan diversos tipos de arritmias, tanto ventriculares como supraventriculares y pueden propiciar MSC. Pueden aparecer como mutaciones nuevas o transmitirse en una familia con diversos grados de penetrancia (número de individuos en una familia con la mutación que expresan el fenotipo) y expresividad (diversos fenotipos que puede presentar una misma mutación) variable de la enfermedad17,18.
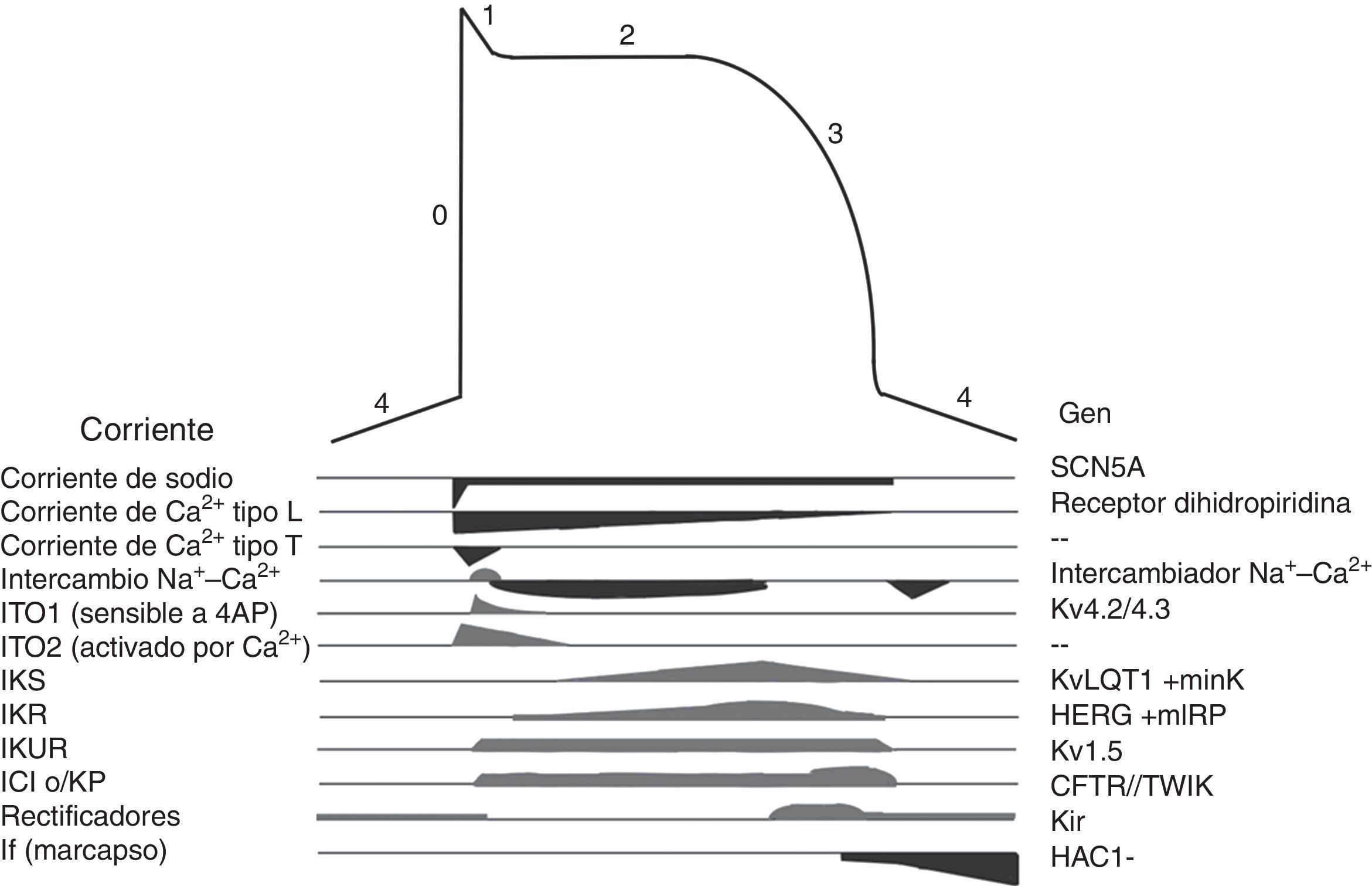
Los canales iónicos son proteínas transmembranales que en la célula miocárdica regulan el movimiento de iones, son responsables de generar el potencial de acción (fig. 3) y de las diferentes morfologías de potencial de acción observadas en las distintas células cardiacas. Las mutaciones en los canales iónicos producen incremento o disminución en determinadas corrientes iónicas lo que altera el potencial de acción con la consecuente irritabilidad celular, lo que condiciona arritmias cardiacas y riesgo de MS. Las canalopatías cardiacas, por lo general, no se acompañan de alteraciones cardiacas estructurales, incluso pueden cursar con un electrocardiograma (ECG) normal y su primera manifestación puede ser la MS19.

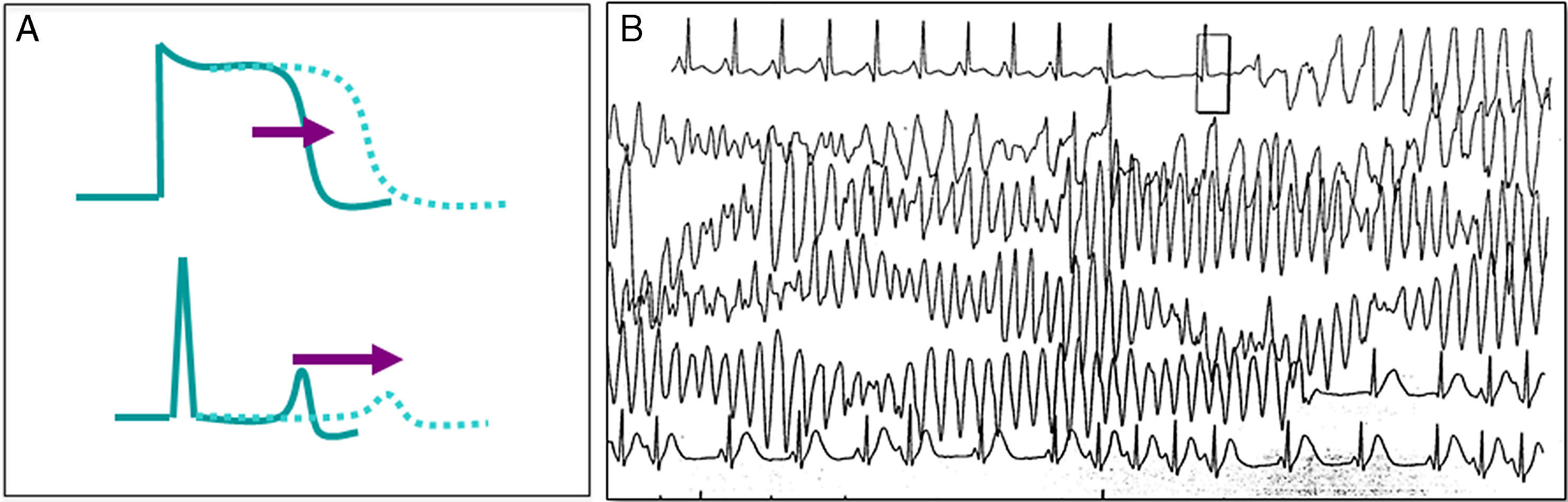
Representa la canalopatía mejor estudiada, se caracteriza por una prolongación en el potencial de acción ventricular que se muestra en el ECG de superficie por un prolongado intervalo QT, lo que favorece arritmias ventriculares del tipo de la taquicardia ventricular polimorfa (fig. 4)20, manifestada por síncope recurrente; en ocasiones incluso puede existir un cuadro similar a crisis convulsivas, lo que puede complicar el diagnóstico o el paciente puede cursar con MS como primera manifestación de la enfermedad. Para el diagnóstico, es importante corregir el intervalo QT a la frecuencia cardiaca, para lo que se puede utilizar la fórmula de Bazett (QTc=√QT medido/RR, en ms)21. Se considera un intervalo QT corregido prolongado aquel>460ms en mujeres o 440ms en hombres medido en DIII o V522, aunque el ultimo consenso internacional en arritmias considera diagnóstico electrocardiográfico de síndrome de QT largo (SQTL) solo en aquellos pacientes con un QTc mayor de 500ms en más de un ECG subsecuente, o bien en pacientes con síncope y QTc entre 480-499 en ausencia de causas secundarias que prolonguen el intervalo QT23. Se puede también diagnosticar SQTL utilizando la escala de puntos desarrollada por Schwartz et al.24, los cuales considera la edad, historia personal, familiar y QTc. Un puntaje≥3.5 es suficiente para hacer el diagnóstico de SQTL23.

El paciente con SQTL puede presentar como principales síntomas el síncope recurrente, crisis convulsivas o MS como primera manifestación de la enfermedad. Hasta la fecha se han descrito 13 genes implicados en la enfermedad (tabla 1), sin embargo hasta en un 30% de los pacientes no se identifica el sustrato genético que explique la enfermedad. A pesar de que se han identificado 12 genes, el 90% de los pacientes con estudio genético positivo presentarán mutaciones en uno de tan solo 3 principales genes:
Genes implicados en el síndrome de QT largo
| Tipo | Gen | Locus | Proteína | Corriente |
| LQT1 | KCNQ1 | 11p15.5 | KvLQT1 | K |
| LQT2 | KCNH2 | 7q35-36 | HERG | K |
| LQT3 | SCN5A | 3p21-24 | Nav1.5 | Na |
| LQT4 | ANK2 | 4q25-27 | Anquirina B | Múltiple |
| LQT5 | KCNE1 | 21q22.1 | MinK | K |
| LQT6 | KCNE2 | 21q22.1 | MiRP | K |
| LQT7, síndrome de Andersen | KCNJ2 | 17q23 | Kir2.1 | K |
| LQT8, síndrome de Timothy | CACNA1c | 12p13.3 | Cav1.2 | Ca |
| LQT9 | CAV3 | 3p25 | CAV3 | |
| LQT10 | SCN4B | 11q23 | SCN4B | Na |
| LQT11 | AKAP9 | 7q21 | AKAP9 | Múltiple |
| LQT12 | SNTA1 | SNTA1 | Na | |
| LQT13 | KCNJ5 | KCNJ5 | IKAch |
1. KCNQ1 que codifica el canal lento de potasio IKs, mutaciones con pérdida en la función de este canal condicionan el SQTL tipo 1 (SQTL1). El canal IKs es sumamente sensible a los estímulos adrenérgicos y la prolongación del intervalo QT puede ser mayor durante o después del ejercicio, al despertar, o al administrar epinefrina. En el SQTL1 se ha reportado que en la infancia (<13 años) los hombres tienen un riesgo mayor de eventos arrítmicos o MS comparado con las mujeres. Después del inicio de la adolescencia, este riesgo se iguala en ambos sexos25. Las mutaciones localizadas en las asas citoplasmáticas del canal IKs se asocian a mayor riesgo de eventos arrítmicos en mujeres, pero no en hombres26. Un valor de intervalo QTc por encima de 500ms representa el factor individual más importante en la predicción de eventos en ambos sexos27. La forma autosómica recesiva del SQTL1 condiciona el síndrome de Jervell-Lange-Nielsen, caracterizado por SQTL, sordera congénita y una elevada incidencia de MS.
2. KCNH2 que codifica el canal rápido de potasio IKr (conocido también como HERG), mutaciones que condicionan pérdida de la función de este canal, generan el SQTL tipo 2. Este canal es también el principal involucrado en la prolongación del intervalo QT inducida por fármacos. Interesante es que los pacientes con SQTL tipo 2 cursan con mayor susceptibilidad de presentar arritmias con estímulos auditivos inesperados (p. ej., reloj despertador)28 o en el periodo posparto (hasta 9 meses después)29,30.
Los pacientes con mutaciones localizadas en el poro del canal IKr tienen un riesgo mayor de eventos arrítmicos que los pacientes con mutaciones en otras regiones del canal31.
3. SCN5A que codifica el canal de sodio isoforma cardiaca NaV1.5 y que condiciona el SQTL tipo 3, que se caracteriza por un incremento en la función del canal, con corriente de sodio (INa) particularmente de tipo tardío, después de la fase 0, afectando las fases 2-3 del potencial de acción. Mutaciones en el canal de sodio que generan SQTL suelen dar manifestaciones muy tempranas, el intervalo QT suele ser>500ms y con frecuencia se observa bloqueo AV 2x1 intermitente32,33 o alternancia eléctrica de la onda T. Los pacientes con SQTL3 suelen presentar eventos arrítmicos durante el reposo o sueño.
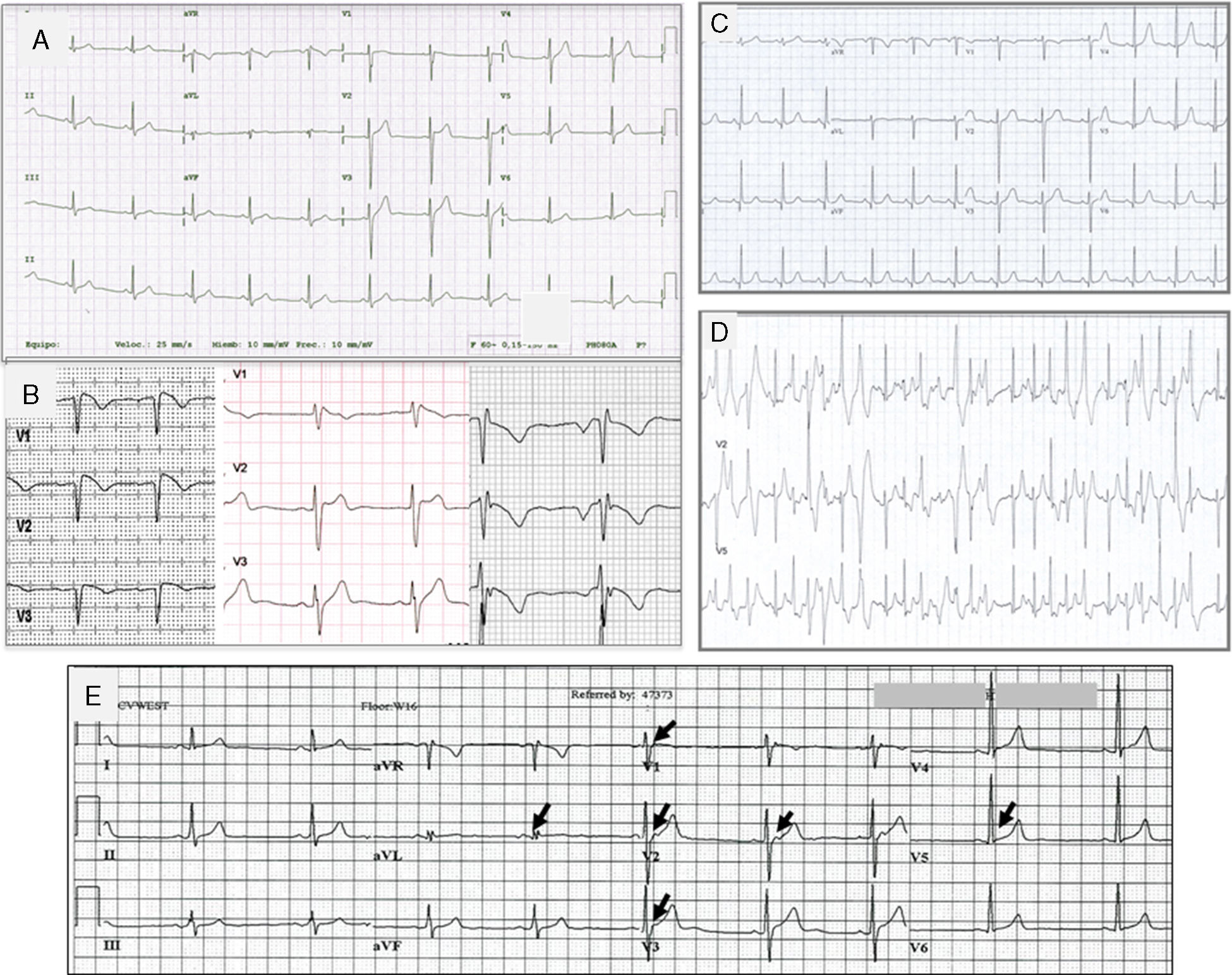
Síndrome de QT cortoEl síndrome de QT corto (SQTC) es una entidad que condiciona un elevado riesgo de MS, se caracteriza por un intervalo QT corregido corto (<330ms), con onda T alta y picuda y el intervalo entre el pico y el final de la onda T no prolongado y alta vulnerabilidad a arritmias ventriculares y MS (fig. 5). El SQTC también se puede diagnosticar si el QTc es<360ms en presencia de una mutación patogénica, antecedentes familiares de SQTC o MS antes de los 40 años, o si se tiene documentado un episodio de taquicardia o fibrilación ventricular en ausencia de enfermedad estructural23. La edad promedio de diagnóstico es de 30 años (4 a 80 años)34. La mayoría de los pacientes con SQTC pueden tener una historia familiar de MS y/o fibrilación auricular. La gravedad de las manifestaciones clínicas del SQTC es muy variable. Los pacientes pueden cursar asintomáticos (38%) o presentar síncope (24%) o palpitaciones (31%) debido a fibrilación auricular o extrasistolia ventricular35.

Trazos electrocardiográficos representantivos de: A. Síndrome de QT corto; B. Síndrome de Brugada. A la izquierda el ECG tipo I, en medio tipo II y a la derecha tipo III; C. Trazo basal en reposo de un paciente con TVPC; D. Trazo del mismo paciente C, al esfuerzo que muestra múltiples extrasístoles polimórficas; E. Paciente de 18 años con fibrilación ventricular y repolarización precoz. Corazón estructuralmente normal. Las flechas indican elevación del punto J, u «onda J».
El SQTC tipo 1 se debe a mutaciones en el gen KCNH2 (HERG)36 que inducen una rápida activación de las corrientes de potasio, esto es, ganancia en la función de IKr y un acortamiento de los potenciales de acción ventriculares. Generalmente, los eventos cardiacos se asocian a situaciones que incrementan el tono adrenérgico
El SQTC tipo 2 ha sido relacionado con mutaciones en el gen KCNQ137 que condicionan una ganancia de función del canal de potasio, lo que da lugar a un acortamiento del potencial de acción. Existe una entidad particular por alteración de este mismo gen que se manifiesta in útero como bradicardia, que en el periodo neonatal se diagnostica de fibrilación auricular y QT corto.
El SQTC tipo 3 presenta mutaciones en el gen KCNJ238 localizado en el cromosoma 17 y generan una ganancia en la función y aceleración de la fase 3 del potencial de acción (tabla 2).
Genes implicados en el síndrome de QT corto
| Tipo | Gen | Locus | Proteína | Corriente |
| SQTS1 | KCNH2 | 7q35 | HERG | IKr |
| SQTS2 | KCNQ1 | 11p15.5 | KvLQT1 | IKs |
| SQTS3 | KCNJ2 | 17q23 | Kir2.1 | IK1 |
| SQTS4 | CACNA1c | Cav1.2 | ICaL | |
| SQTS5 | CACNB2 | Subunidad beta 2 del canal de calcio tipo L | ICaL | |
| SQTS6 | CACNA2D1 | Subunidad gamma 1 de canal de calcio tipo L | ICaL |
El síndrome de Brugada (SBr) se describió originalmente como un desorden arrítmico con herencia autosómica dominante, caracterizado por elevación del ST y onda T negativa en las precordiales derechas sin anomalías cardiacas estructurales (fig. 5)39. Se calcula que la prevalencia del SBr se sitúa en torno a 5/10,000 habitantes40.
Los pacientes con SBr permanecen en su mayoría asintomáticos. No obstante, se ha descrito que un 17-42% de ellos presentan síncope o MS como consecuencia de una arritmia ventricular en algún momento de su vida. Este síndrome es mucho más frecuente en varones que en mujeres41. Aunque la media de edad al inicio de los eventos es alrededor de los 40 años, la MS puede afectar a personas de todas las edades, especialmente varones (75%).
Se han descrito 3 patrones electrocardiográficos distintos (fig. 5B):
El patrón tipo i, caracterizado por una elevación descendente del segmento ST de 2mm en al menos una derivación precordial derecha (V1-V3), seguida de ondas T negativas. El patrón tipo ii, caracterizado por elevación del segmento ST 2mm en precordiales derechas seguida de ondas T positivas o isodifásicas, lo que confiere al ECG un aspecto de silla de montar. El patrón tipo iii, definido como cualquiera de los 2 anteriores si la elevación del segmento ST es≤1mm. Los 3 patrones pueden observarse en el SBr, incluso en el mismo paciente en momentos diferentes. Sin embargo solo el tipo i se considera diagnóstico de la enfermedad. En el nuevo consenso internacional, se considera diagnóstico de SBr en aquellos pacientes con ECG tipo i en al menos una derivación precordial (V1 o V2), en posición estándar o posicionados en el segundo espacio intercostal, espontáneo o inducido con fármacos23.
En algunos pacientes, las alteraciones electrocardiográficas son transitorias y suelen ser más evidentes al administrarse bloqueadores de los canales de Na, tales como flecainida, ajmalina o procainamida42. La flecainida puede inducir además prolongación del QT que se observa sobre todo en las derivaciones precordiales derechas (V1-V2)43. Otros medicamentos, tales como los betabloqueadores, antidepresivos tricíclicos, tetracíclicos, litio y anestésicos locales pueden inducir la manifestación eléctrica del síndrome. Eventos clínicos tales como fiebre, hipopotasemia, hiperpotasemia, hipercalcemia, intoxicación con alcohol o cocaína; la estimulación cardiaca, maniobras vagales y el tono adrenérgico elevado también pueden evidenciar o modular el patrón electrocardiográfico del SBr44. Para mayor información sobre la lista de fármacos que deben ser evitados en el SBr, consultar http://brugadadrogs.org.
De los pacientes afectados, un 20-50% tiene antecedentes de MS familiar. El SBr es una enfermedad de herencia familiar autosómica dominante y penetrancia variable.
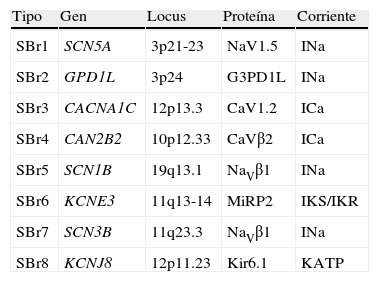
Las primeras mutaciones en el SBr fueron halladas el gen SCN5A, que codifica para el canal de sodio cardiaco; dichas mutaciones condicionan pérdida de la función del canal con disminución de la corriente. Solo un 18-30% de los pacientes tienen mutaciones en este gen, de hecho se han identificado hasta la fecha 12 genes asociados al SBr (tabla 3), los tipos 2-12 explican tan solo el 5% de los pacientes45.
Genes implicados en el síndrome de Brugada
| Tipo | Gen | Locus | Proteína | Corriente |
| SBr1 | SCN5A | 3p21-23 | NaV1.5 | INa |
| SBr2 | GPD1L | 3p24 | G3PD1L | INa |
| SBr3 | CACNA1C | 12p13.3 | CaV1.2 | ICa |
| SBr4 | CAN2B2 | 10p12.33 | CaVβ2 | ICa |
| SBr5 | SCN1B | 19q13.1 | NaVβ1 | INa |
| SBr6 | KCNE3 | 11q13-14 | MiRP2 | IKS/IKR |
| SBr7 | SCN3B | 11q23.3 | NaVβ1 | INa |
| SBr8 | KCNJ8 | 12p11.23 | Kir6.1 | KATP |
SBr: síndrome de Brugada.
En pacientes con claro diagnóstico de SBr, la tasa de eventos cardiacos por año en aquellos con MS abortada es del 7.7%, en aquellos con síncope únicamente es del 1.9% y del 0.5% en los pacientes asintomáticos46.
Taquicardia ventricular polimorfa catecolaminérgicaLa taquicardia ventricular polimorfa catecolaminérgica (TVPC) es un trastorno del ritmo potencialmente letal y heredable, caracterizado por arritmias ventriculares al estrés o al estímulo adrenérgico que provocan síncope o muerte súbita, con una tasa de mortalidad del 35-50% a la edad de 35 años, por lo que constituye una de las canalopatías cardiacas más graves. El corazón suele ser estructuralmente normal y por lo regular el ECG en reposo no muestra alteraciones, pero durante el ejercicio es común que se desencadenen extrasístoles ventriculares polimorfas (figs. 5C y 5D), o bien taquicardia ventricular polimorfa o bidireccional que pueden degenerar en fibrilación ventricular. Así, el síntoma inicial de la TVPC es síncope o MS durante el ejercicio47. Es así que la TVPC puede diagnosticarse cuando existe taquicardia ventricular bidireccional o polimorfa inducida por catecolaminas o ejercicio en el contexto de un corazón estructuralmente normal, en individuos menores de 40 años de edad o en mayores de 40 años de edad cuando se ha descartado enfermedad coronaria. Puede también diagnosticarse si se detecta una mutación implicada en la enfermedad.
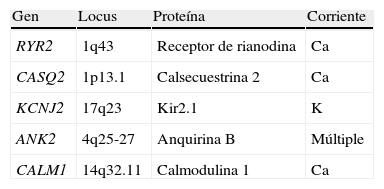
Se han identificado 2 variantes genéticas, la autosómica dominante causada por mutaciones en el gen del receptor de la rianodina RyR2 (1q42-Q43)48, y la recesiva, causada por mutación en la isoforma del gen de la calsecuestrina (CASQ2)49. Ambos genes están implicados en la regulación del calcio intracelular y ambos defectos generan un incremento en la salida de calcio del retículo sarcoplásmico. Este exceso de calcio se relaciona con alteraciones en el potencial de membrana del sarcolema, con aparición de despolarizaciones tardías que facilitan las arritmias. El receptor de la rianodina es un canal de calcio intracelular que se encuentra en el retículo sarcoplásmico y se activa por la entrada de pequeñas cantidades de calcio, permitiendo la salida del calcio almacenado. Se han identificado más de 70 mutaciones en RyR2. La calsecuestrina es también una proteína del retículo sarcoplásmico, con gran afinidad al ion calcio y que funciona como «almacén» de este ion.
Existen otros genes que pueden dar «fenocopias» de TVPC y que de hecho han sido asociados a otras canalopatías, como KCNJ2 que da el Sx. de Andersen o SQTL7 y ANKB descrito inicialmente en SQTL4. Asmismo, recientemente se han identificado mutaciones en el gen CALM1 que codifica la calmodulina-cinasa en pacientes con TVPC (tabla 4)50.
Síndromes de repolarización precoz y fibrilación ventricular idiopáticaEl patrón electrocardiográfico de repolarización precoz se caracteriza por un ascenso del punto J (la unión entre el final del complejo QRS y el inicio del segmento ST) en derivaciones diferentes a V1-V3; es un hallazgo relativamente frecuente y clásicamente se ha considerado benigno sin repercusión clínica. Sin embargo, estudios recientes han confirmado su asociación con fibrilación ventricular idiopática.
Se diagnostica síndrome de repolarización precoz cuando se observa elevación del punto J≥1mm en más de 2 derivaciones contiguas inferiores o laterales en pacientes que han sobrevivido o no a un episodio de fibrilación ventricular idiopática23.
El mecanismo por el que se relaciona el patrón electrocardiográfico y la susceptibilidad a arritmias ventriculares es desconocido. Se ha sugerido que la elevación del punto J sería un marcador de la presencia de cierto grado de heterogeneidad de la repolarización ventricular que haría vulnerable al miocardio para desarrollar arritmias ante disparadores como un evento isquémico51 (tabla 5).
Prevención primaria y secundaria de la muerte súbita cardiacaDebido a que las canalopatías se caracterizan por la ausencia de cardiopatía estructural, la indicación de un desfibrilador automático implantable (DAI) es clase i en todos los pacientes que hayan tenido un episodio de muerte súbita (prevención secundaria), independientemente de la fracción de eyección del ventrículo izquierdo. Sin embargo, la indicación del DAI en prevención primaria es controvertida y deberá ser evaluada en cada caso en particular.
En los pacientes de SQTL como medida general se debe indicar cambios en el estilo de vida, esto es, evitar ejercicio extenuante, particularmente nadar en el SQTL1. Reducción a exposición de ruidos abruptos altos en SQTL tipo 2 y evitar fármacos que prolongan el intervalo QT en todos los pacientes con SQTL. La terapia farmacológica con betabloqueadores es efectiva para reducir el riesgo de eventos cardiacos. Es por esto que los betabloqueadores tienen una indicación clase i en todos los pacientes asintomáticos con diagnóstico de SQTL52. De acuerdo al registro internacional de SQTL, la efectividad de los betabloqueadores en la prevención de síntomas arrítmicos y muerte, varía de acuerdo al tipo, siendo del 81% para el tipo 1, del 59% para el tipo 2 y del 50% para el tipo 3. Existen diversos fármacos con efecto betabloqueador, pero particularmente 2 son útiles en el tratamiento del SQTL: el propranolol y el nadolol. El tratamiento con metoprolol ha demostrado menor efecto protector en el contexto del SQTL53. En el SQTL tipo 3, los antiarrítmicos como la mexiletina y la flecainida han mostrado utilidad y tienen una indicación iia en pacientes con QTc>500ms en quienes el tratamiento inicial logre reducir en 40ms el QTc23. En los pacientes con SQTL «oculto» es decir, con una mutación predisponente de SQTL pero con QT normal, los betabloqueadores tienen una indicación iia. Los pacientes con SQTL que han presentado síncope o taquicardia ventricular a pesar del tratamiento con betabloqueadores, presentan un riesgo 3.6 veces mayor de presentar arritmias malignas54 y tienen una indicación de DAI iia. La denervación simpática izquierda tiene indicación clase i en aquellos pacientes en los que a pesar de que se ha indicado DAI este no se pueda implantar o en aquellos pacientes a los que no se puedan administrar betabloqueadores. Tendrá indicación iia en los pacientes con SQTL sintomáticos a pesar del tratamiento con betabloqueadores o DAI23. El DAI no está indicado (clase iii) en aquellos pacientes con SQTL asintomático en quienes no se haya intentado tratamiento con betabloqueadores.
En los pacientes con SQTC, existe poca información clínica, se considera de utilidad el tratamiento con quinidina en los pacientes con mutaciones en KCNH2. Sin embargo se desconoce si hay una reducción real de la mortalidad con este fármaco. Es importante mencionar que no existen estudios con adecuado número de pacientes que permitan normar conducta en esta enfermedad, sin embargo, la quinidina y la disopiramida en series pequeñas han demostrado prolongar el intervalo QT al rango normal (SQTC1)47.
El DAI debe considerarse en todos los pacientes sintomáticos con SQTC.
En el SBr, el tratamiento farmacológico no es tan efectivo; la quinidina ha mostrado ser de utilidad en los pacientes sintomáticos55, pero se desconoce el papel que desempeña en la prevención primaria de arritmias ventriculares en pacientes asintomáticos con SBr. Por otro lado, se ha reportado que los pacientes con SBr tienen una incidencia mayor de arritmias supraventriculares, particularmente fibrilación auricular. En este escenario resulta complejo el tratamiento médico pues fármacos como la flecainida o cualquier otro bloqueador de canal de sodio se encuentra contraindicado. La amiodarona también ha mostrado tener un efecto proarrítmico mayor en los pacientes con SBr. En el contexto de arritmias supraventriculares, la quinidina puede ser una buena opción terapéutica. El isoproterenol es un fármaco que se ha utilizado con éxito para el tratamiento agudo de tormentas eléctricas en el SBr56.
Es importante notar que los diversos estudios multicéntricos han demostrado que la tasa de mortalidad es en realidad baja en los pacientes asintomáticos, como mencionamos anteriormente; el desfibrilador debe indicarse únicamente en los pacientes considerados de alto riesgo. No existe duda de que los pacientes con mayor riesgo son aquellos con síntomas y la presencia espontánea de SBr tipo 1 en el ECG. Sin embargo, se considera que los siguientes factores se asocian a mayor riesgo de arritmias en el SBr: fragmentación del QRS57, periodo refractario efectivo ventricular<200ms58, género masculino59 y fibrilación auricular60. La historia familiar de MS, la inducibilidad de arritmias ventriculares durante el estudio electrofisiológico y la presencia de una mutación en SCN5A, no han mostrado ser predictores de mayor riesgo46. Sin embargo, algunas mutaciones específicas como las que resultan en una proteína truncada, pueden asociarse a fenotipos más severos61.
En el tratamiento del SBr se considera indicación i el implante del DAI en prevención secundaria de MS en pacientes sintomáticos con ECG tipo i En pacientes con SBr espontáneo en el ECG y síncope únicamente y pacientes en quienes se ha documentado taquicardia ventricular sin historia de muerte súbita, la implantación del DAI es iia23. Si se ha inducido fibrilación ventricular durante el estudio electrofisiológico en un caso con SBr asintomático, el DAI tendrá una indicación iib. El tratamiento farmacológico con quinidina en el SBr tiene un futuro prometedor, pero actualmente es una indicación iib.
En el tratamiento de la TVPC, como medidas generales, es importante evitar ejercicio extenuante o competitivo, así como ambientes que incrementen el estrés. Todos los pacientes tienen indicación de betabloqueador. La flecainida ha mostrado ser útil en la reducción de eventos arrítmicos62,63 así como el verapamilo. Sin embargo, solo los betabloqueadores tienen una indicación clase i23 y la asociación de flecainida con betabloqueadores se ha reportado como útil en pacientes muy sintomáticos.
Los pacientes asintomáticos portadores de una mutación pueden ser tratados con betabloqueador con indicación clase iia23. Se puede considerar la realización de denervación simpática izquierda en aquellos pacientes muy sintomáticos con terapias apropiadas frecuentes por el DAI a pesar del uso de betabloqueadores o en pacientes a los que no se pueda administrar este último fármaco, con indicación iib. El DAI tendrá una indicación clase i en todos aquellos individuos con TVPC sintomáticos a pesar del tratamiento con betabloqueadores o denervación simpática izquierda.
En el tratamiento de los síndromes de repolarización precoz existe consenso de que todo caso sintomático que haya sobrevivido a un episodio de fibrilación ventricular debe recibir un DAI. El tratamiento farmacológico de los episodios de tormenta eléctrica con quinidina y/o isoproterenol, tiene una indicación iia. El DAI está contraindicado en los pacientes asintomáticos con repolarización precoz23.
Estudios genético-molecularesEn todo paciente en el que se sospeche una canalopatía arritmogénica debe realizarse idealmente el estudio genético. La principal utilidad en todas las canalopatías es el diagnóstico en cascada que se puede hacer de familiares que podrían estar en riesgo de padecer también la misma enfermedad. El estudio genético tendrá implicaciones terapéuticas sobre todo en el SQTL donde el tipo de mutación puede orientar al tratamiento médico como se indicó previamente. Finalmente, el estudio genético tiene también implicaciones pronósticas; esto se ha visto principalmente en SQTL y el SBr donde la localización y/o tipo de mutación puede pronosticar la gravedad del fenotipo.
El poder detectar una mutación en un individuo también lo hace candidato a poder utilizar técnicas de fertilización in vitro usando células preseleccionadas que no contengan la mutación para así evitar la transmisión genética de la enfermedad a la descendencia.
Sin embargo es importante tener en cuenta que el estudio genético no siempre detecta el defecto en los pacientes afectados, a pesar de que el fenotipo sea claro. La posibilidad de detectar mutaciones en el SQTL es del 70%, en SQTC se desconoce el número exacto, en SBr es del 40%, en TVPC del 50% y en el síndrome de repolarización precoz está cerca del 30%, por lo que aún se llevan a cabo estudios para determinar causas genéticas de estas enfermedades.
En todo paciente en que se solicita estudio genético se debe proporcionar adicionalmente una consulta genética para explicar con claridad los resultados, que en ocasiones suelen ser complejos, particularmente cuando varios genes podrían estar implicados en el fenotipo o cuando existe penetrancia incompleta o variación del fenotipo.
Si bien los estudios genéticos han resultado tradicionalmente caros y generalmente fuera del alcance de los países en desarrollo, las técnicas han mejorado con el tiempo, siendo en la actualidad más efectivas, baratas y rápidas, lo que promete un acceso fácil para la población; particularmente con las técnicas de secuenciación de la siguiente generación64. En los años recientes se ha multiplicado la posibilidad de efectuar la lectura de todos los exones de un individuo (análisis de exoma) lo que seguramente revolucionará el uso de marcadores genéticos para el diagnóstico, pronóstico e incluso tratamiento oportuno de muchas enfermedades, incluyendo las canalopatías cardiacas. Este abordaje ya empieza a ser una realidad y en varios centros hospitalarios se utiliza hoy en día como herramienta diagnóstica de enfermedades en donde existe contribución genética.
Los estudios funcionales de las diversas mutaciones encontradas han ayudado también a entender importantes aspectos fisiológicos de los diversos canales y a delimitar regiones críticas que al ser alteradas por alguna mutación afectan su funcionamiento. Dichos estudios no están siempre al alcance del clínico pero representan una herramienta importante en la clasificación de mutaciones.
Asimismo, en los años recientes, se han desarrollado diversas técnicas que contemplan la obtención de cardiomiocitos a partir de fibroblastos obtenidos a través de una biopsia de piel. Este tipo de estudios ofrece la posibilidad de practicar una medicina individualizada y quizá en un futuro se puedan utilizar en la práctica clínica cotidiana65.
ConclusionesLa MSC se debe a un grupo heterogéneo de enfermedades. Una vez descartada la presencia de cardiopatía estructural, las canalopatías cardiacas suelen ser una de las principales causas. Explican el 30% de los pacientes con MS inexplicada y el 10% de los pacientes con MS infantil. El estudio genético o autopsia molecular es de gran utilidad y podrá ayudar a detectar otros miembros de la familia potencialmente afectados.
FinanciaciónEste trabajo ha sido realizado gracias al financiamiento otorgado por el CONACyT.
Conflicto de InteresesLos autores declaran no tener ningún conflicto de intereses.
Se agradece a CONACyT el fondo otorgado a los autores para el desarrollo de una plataforma, que permitirá el estudio genético de los pacientes con muerte súbita a un precio razonable en México.