





El complejo de Carney es un síndrome de neoplasia múltiple de tumores endocrinos y no endocrinos, que incluye la presencia de mixoma, lentiginosis cutánea y enfermedad nodular primaria pigmentada, entre otros criterios para el diagnóstico.
En la mayoría de los casos es de transmisión autosómica dominante, por lo que su diagnóstico hace necesario el estudio y seguimiento familiar. Se ha identificado la presencia de mutaciones inactivantes del gen PRKAR1A como causante de la enfermedad. Desde el año 2015 se han agregado otros genes relacionados, como variantes activantes del gen PRKACA y PRKACB.
En este trabajo se ahondará en los aspectos genéticos relacionadas con el complejo de Carney.
Carney complex is a multiple neoplasia syndrome having endocrine and non-endocrine manifestations. Diagnostic criteria include myxoma, lentigines, and primary pigmented nodular adrenocortical disease, amongst other signs/symptoms.
In most cases it is an autosomal dominant disease, and diagnosis therefore requires study and follow-up of the family members. Inactivating mutations of the PRKAR1A gene were identified as the main cause of the disease, although since 2015 other disease-related genes, including PRKACA and PRKACB activating mutations, have also been related with Carney complex.
This review will address the genetic aspects related to Carney complex.
El complejo de Carney (CNC, OMIM 160980) fue inicialmente descrito en 1985 por J. Aidan Carney, como la asociación de mixoma, lesiones cutáneas pigmentadas e hiperactividad de ejes endocrinos1,2. Se trata de un síndrome de neoplasia múltiple con presencia de tumores endocrinos y no endocrinos. En el 70% de los casos es transmitido siguiendo un patrón de herencia autosómica dominante con penetrancia completa, mientras que en el resto es de presentación esporádica3–5.
La prevalencia del CNC se desconoce hasta el momento debido a su baja frecuencia. En la cohorte internacional más grande se han descrito aproximadamente 750 casos5,6.
La principal manifestación endocrina con que se relaciona al CNC es la hiperplasia adrenal nodular primaria pigmentada (PPNAD por sus siglas en inglés)7,8. Esta es una causa de síndrome de Cushing independiente de ACTH9,10.
En el año 2000 se describió que el CNC es causado por mutaciones con pérdida de función o grandes deleciones del gen PRKAR1A (OMIM 188830) localizado en el cromosoma 17q 22_24, que codifica a la subunidad regulatoria alfa i de la proteína cinasa A11. También se encontró un segundo locus del CNC ubicado en el cromosoma 2p1612. Más recientemente se han publicado otras mutaciones, incluyendo defectos en los genes PRKACA y PRKAC9.
Desde la descripción inicial del CNC y la identificación de las mutaciones clásicamente relacionadas con la enfermedad, se han publicado variantes genéticas y clínicas de la misma.
Aspectos molecularesLos mecanismos moleculares del CNC están relacionados con amplificación de la señal del adenosín monofosfato cíclico (AMPc), por mutaciones en componentes de su vía intracelular11.
El AMPc actúa induciendo una cascada de fosforilaciones que determinan modificaciones en proteínas y en la transcripción de genes. Está localizado en todo el cuerpo humano y es esencial para regular numerosas funciones biológicas. Su activación por la subunidad alfa (Gα) de receptores de membrana celular acoplados a proteína G desencadena el inicio de su actividad en las células.
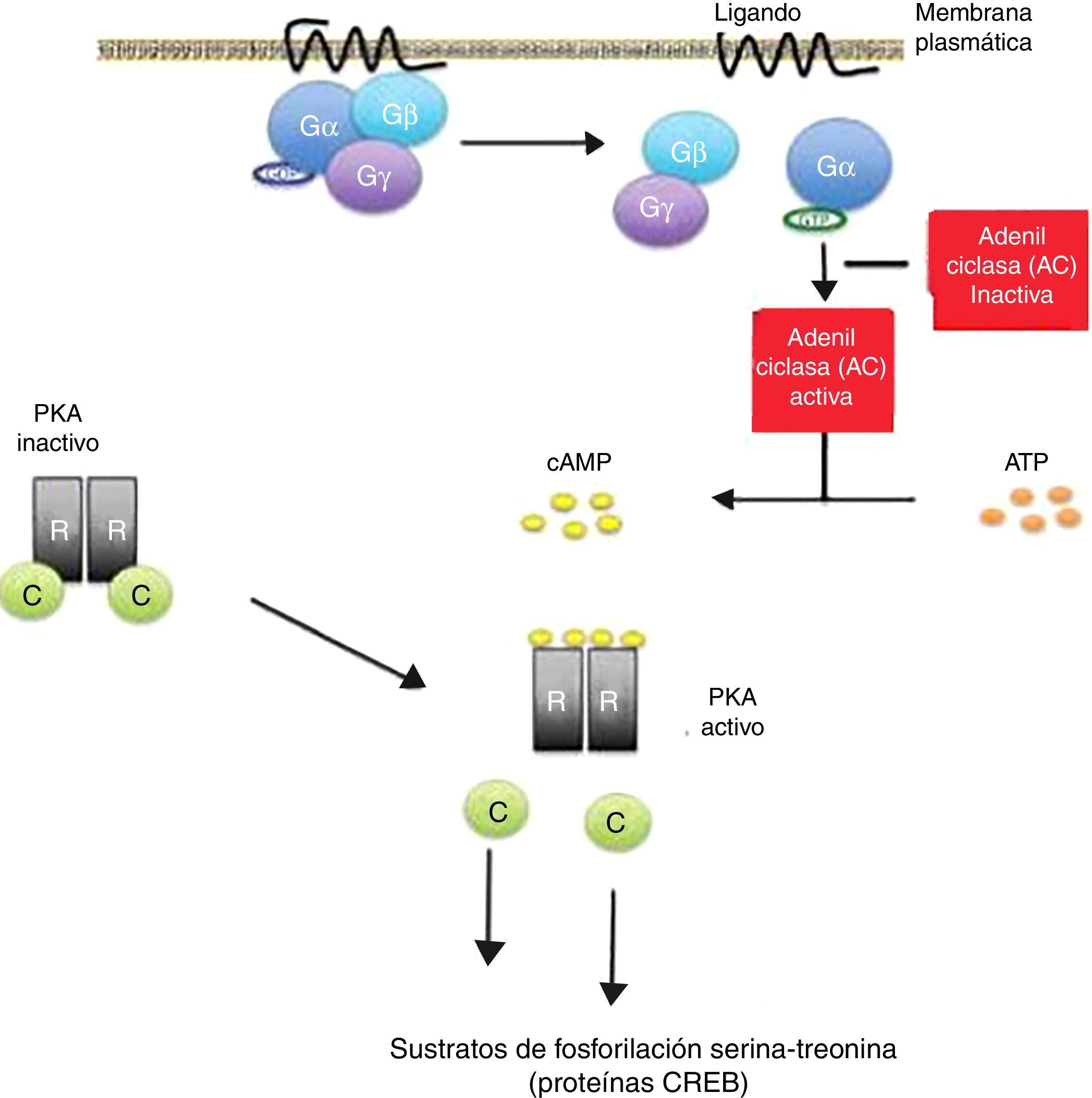
La fijación de ciertos ligandos o «primeros mensajeros» al receptor de membrana asociado a proteína Gs conduce a un aumento del AMPc, que actúa como un «segundo mensajero» (fig. 1). Este último induce diversos cambios en el metabolismo celular que difieren en los distintos tipos de células13,14. El final de la respuesta inducida por AMPc es a través de las enzimas fosfodiesterasas, que actúan hidrolizando a esta molécula15,16.
: La activación del receptor de proteína G por unión de un ligando estimula la enzima adenilato ciclasa (AC) para la síntesis de AMPc. Todos los genes regulados por AMPc contienen una secuencia de ADN de acción cis, llamado elemento de respuesta al AMPc (CRE), que fija la forma fosforilada de un factor de transcripción denominado proteína de unión CRE (CREB). La liberación de las subunidades catalíticas (C) del complejo proteína quinasa A (PKA) se traslocan al núcleo y fosforilan a las proteínas CREB en los sustratos de fosforilación serina-treonina, en respuesta a niveles aumentados de AMPc. Las mutaciones inactivantes del gen PRKAR1A en el complejo de Carney resultan en amplificación de la señal del AMPc.")
Vía de activación del adenosín monofosfato cíclico (AMPc): La activación del receptor de proteína G por unión de un ligando estimula la enzima adenilato ciclasa (AC) para la síntesis de AMPc. Todos los genes regulados por AMPc contienen una secuencia de ADN de acción cis, llamado elemento de respuesta al AMPc (CRE), que fija la forma fosforilada de un factor de transcripción denominado proteína de unión CRE (CREB).
La liberación de las subunidades catalíticas (C) del complejo proteína quinasa A (PKA) se traslocan al núcleo y fosforilan a las proteínas CREB en los sustratos de fosforilación serina-treonina, en respuesta a niveles aumentados de AMPc.
Las mutaciones inactivantes del gen PRKAR1A en el complejo de Carney resultan en amplificación de la señal del AMPc.
El complejo enzimático proteína cinasa A (PKA por sus siglas en inglés) es el receptor intracelular más importante del AMPc en las células eucariotas. La PKA modula la actividad de varias proteínas, fosforilando el grupo hidroxilo en los residuos serina y/o treonina de varias proteínas, por lo que se trata de una serina/treonina proteína cinasa específica17.
En estado inactivo, la PKA es un complejo tetramérico que consiste en 2 subunidades reguladoras (R): tipo i-α (RI-α) y tipo ii-β (RII-β) y 2 subunidades catalíticas (C): tipo i-α (CI-α) y tipo ii-β (CII-β). Es decir, se trata de un dímero de subunidades reguladoras unidas a 2 subunidades catalíticas. Cada subunidad R tiene 2 sitios de fijación al AMPc separados. La fijación del AMPc a ambos sitios en una subunidad R conduce a la liberación de la subunidad C asociada, desenmascarando su sitio catalítico y activando su actividad cinasa18. La función más conocida que se le atribuye a las subunidades R es la inhibición de la actividad cinasa que poseen las subunidades C19.
La fijación del AMPc a una subunidad R sucede de modo cooperativo, es decir, la unión de la primera molécula de AMPc disminuye la constante de velocidad de la reacción directa (Kd) para la fijación de la segunda. Así, pequeños cambios en el nivel de AMPc citosólico pueden causar grandes cambios en la cantidad de subunidades catalíticas disociadas y consecuentemente, en la actividad cinasa del complejo14,20.
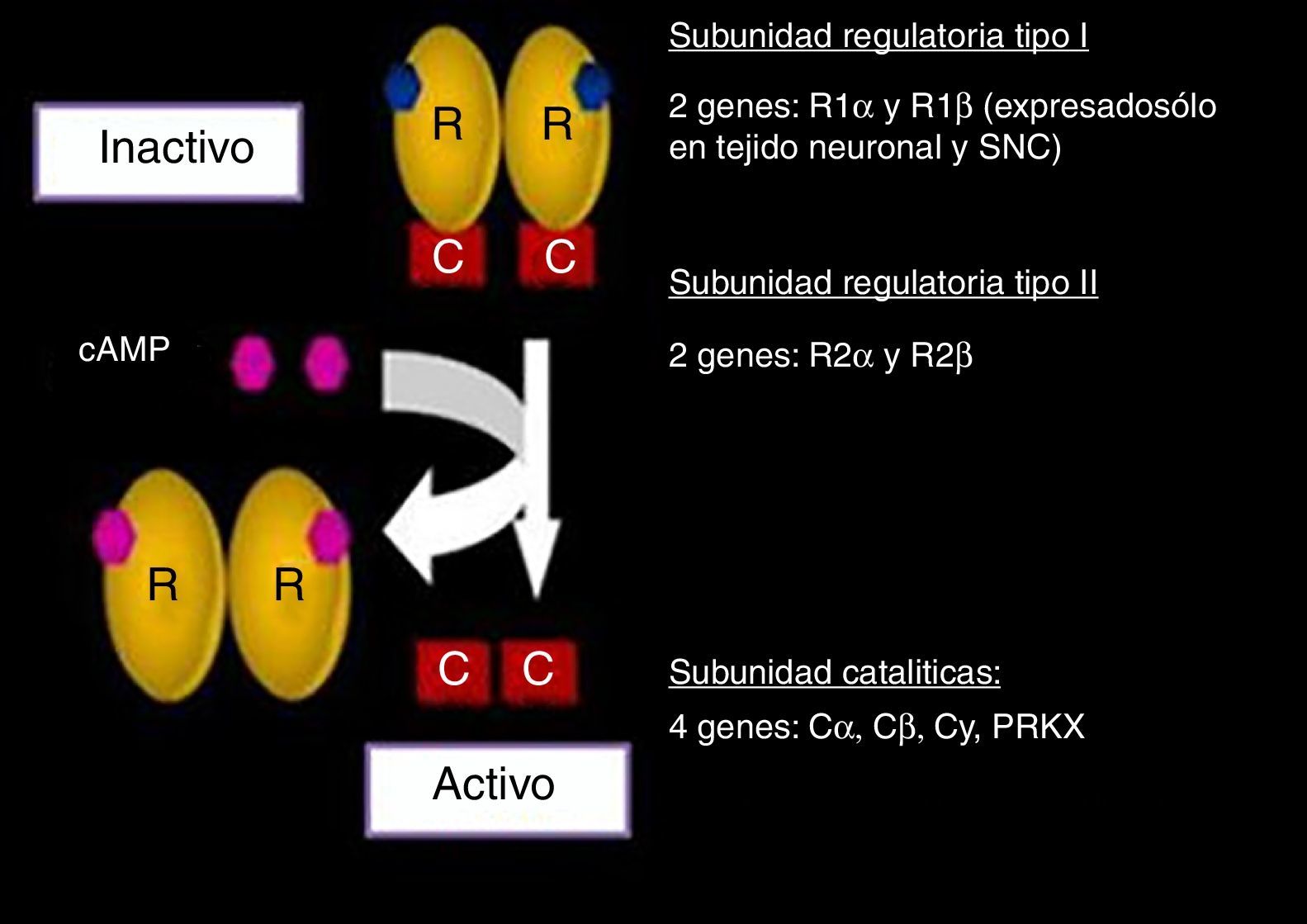
Se estudiaron los genes que codifican para cada una de las subunidades que conforman el complejo PKA (fig. 2). La subunidad RI se encuentra ampliamente en todo el organismo codificado por el gen PRKAR1A o gen de la subunidad reguladora 1A de la proteína cinasa A. En tejido neuronal esta subunidad está codificada por R1 β, expresado únicamente en dichos tejidos11,12,19.
 y genes relacionados. Modificado y publicado con permiso de Stratakis22.")
Complejo proteína cinasa A (PKA) y genes relacionados. Modificado y publicado con permiso de Stratakis22.
En cuanto a la subunidad RII, su expresión está mediada por los genes R2 α y R2 β. Por otra parte, las 2 subunidades catalíticas están reguladas por 4 genes: Ca, Cb, Cy, PRKX. Cada uno de estos ha sido ampliamente estudiado, tanto en su expresión normal como mutada18–25.
Activación de la transcripción genéticaTodos los genes regulados por AMPc contienen una secuencia de ADN de acción cis, llamado elemento de respuesta al AMPc (CRE), que fija la forma fosforilada de un factor de transcripción denominado proteína de unión CRE (CREB), que se encuentra solo en el núcleo de las células. Cuando se produce la liberación de las subunidades C de PKA en respuesta a niveles aumentados de AMPc, estas se traslocan al núcleo y fosforilan a las proteínas CREB en la serina-133. Las proteínas CREB fosforiladas se unen a los genes diana que contienen CRE y también interactúan con un coactivador denominado CBP/300, que enlaza CREB a la maquinaria transcripcional basal y permite que estimule la transcripción del ADN14,21.
La PKA tiene una amplia participación en procesos celulares, incluyendo la transcripción, el metabolismo, la progresión del ciclo celular y la apoptosis. En condiciones normales de inactivación, es decir sin la unión del AMPc, las 4 subunidades de la PKA se encuentran asociadas conformando un complejo tetramérico en estado inactivo. Cuando existe una reducción de un 50% de la función reguladora de la subunidad R1, se origina un incremento en la cascada de fosforilaciones inducidas por PKA, R1 libera su acción reguladora sobre las subunidades C y por consiguiente se produce una falta en el control de la actividad cinasa que poseen estas últimas20–27. De esta forma la inactivación funcional del gen PRKAR1A está asociada con exceso en la vía de señalización de la PKA en los tejidos afectados.
El estudio de la función de la vía del AMPc-PKA y de los genes que la codifican fue esencial para comprender la patogenia y los hallazgos genéticos de los pacientes con CNC. Por otra parte, estudios más recientes involucran esta vía en otras causas de síndrome de Cushing de origen adrenal, desde la PPNAD y el carcinoma adrenal hasta adenomas unilaterales productores de cortisol28–32.
Manifestaciones clínicas del complejo de CarneyEste síndrome se caracteriza por la presencia de tumores de glándulas endocrinas y de otros órganos no endocrinos.
Los criterios diagnósticos fueron revisados en el 2001; en el año 2015 se describieron alteraciones en otros genes aparte del PRKAR1A (tabla 1).
Criterios diagnósticos mayores del complejo de Carney
| 1. Lentiginosis cutánea con distribución típica (labios, conjuntivas, mucosas)a |
| 2. Mixomas (cutáneos y mucosos)a o mixoma cardiacoa |
| 3. Mixomatosis mamariaa o hallazgos en RMN (con supresión grasa) sugestiva del diagnóstico |
| 4. Enfermedad nodular primaria pigmentadaa o aumento paradojal en la excreción de glucocorticoides urinarios tras la administración de dexametasona |
| 5. Acromegalia asociada con adenoma hipofisario productor de GHa |
| 6. Tumor testicular de células grandes calcificadas de Sertolia o presencia de calcificaciones en ecografía testicular |
| 7. Carcinoma de tiroidesa o presencia de múltiples nódulos hipoecoicos en ecografía tiroidea en edad prepuberal |
| 8. Schwannomas melanocíticos psamomatososa |
| 9. Nevus azul, múltiples nevus azules epitelioidesa |
| 10. Múltiples adenomas mamarios ductalesa |
| 11. Osteocondromixomaa |
| Criterios suplementarios |
| 1. Familiar de primer grado afectado |
| 2. Presencia de mutaciones inactivantes del gen PRKAR1A (4) |
| 3. Variantes activantes del gen PRKACA o del PRKACB (9,30,50) |
| Hallazgos sugestivos |
| 1. Múltiples lesiones pigmentadas sin distribución típica |
| 2. Múltiples nevus azules |
| 3. Manchas café con leche u otra desde el nacimiento |
| 4. Aumento de factor de crecimiento insulínico tipo 1, test de SOG para GH anormal o aumento paradojal de GH con el test de TRH, aún sin hallazgos clínicos de acromegalia |
| 5. Cardiomiopatía |
| 6. Antecedente de familiares con síndrome de Cushing, acromegalia o muerte súbita |
| 7. Quiste pilonidal |
| 8. Pólipos colónicos (con frecuencia asociado a acromegalia) |
| 9. Múltiples lunares en piel u otras manifestaciones cutáneas pigmentadas, lipomas múltiples |
| 10. Hiperprolactinemia (con frecuencia leve y combinada con acromegalia clínica o subclínica) |
| 11. Nódulo tiroideo aislado en joven menor de 18 años, múltiples nódulos tiroideos en paciente mayor de 18 años (hallazgo ecográfico) |
| 12. Antecedente familiar de carcinoma, en particular de tiroides, colon, páncreas y ovario; otros tumores múltiples benignos o malignos |
El diagnóstico se establece por la presencia de 2 o más criterios clínicos mayores. Si el paciente tiene una mutación germinal del PRKAR1A y/o un familiar de primer grado afectado con CNC, una sola manifestación clínica es suficiente para el diagnóstico3,4,9.
La lentiginosis en piel y mucosas es la presentación más común del CNC; típicamente incrementa su número durante la pubertad. Presenta una distribución característica alrededor de labios, conjuntivas, mucosa oral y genital33.
En relación con la presencia de mixomas cardiacos, estos ocurren a temprana edad en cualquier cámara cardiaca. Se manifiestan por obstrucción del flujo sanguíneo intracardiaco, fenómenos embólicos y/o insuficiencia cardiaca. Otras localizaciones de mixomas incluyen piel, mama, orofaringe y tracto genital femenino9.
La PPNAD es causa de síndrome de Cushing de origen adrenal y es el tumor endocrino más frecuente relacionado con el CNC, presente en el 25% de los casos. Es sugestivo de esta entidad el aumento paradojal de los niveles de cortisol libre urinario en respuesta a la administración de dexametasona29,34. La PPNAD continúa siendo un desafío diagnóstico por estudios de imagen debido a la apariencia normal de la glándula adrenal y el pequeño tamaño de los nódulos; la confirmación de la entidad es histológica9.
Más de 3 cuartas partes de los hombres con CNC pueden presentar un tumor testicular de células grandes calcificadas de Sertoli. Estos generalmente son bilaterales y multifocales9.
Por otra parte, más de un 75% de los individuos con CNC presentan múltiples nódulos tiroideos, la mayoría relacionados con adenomas foliculares.
Respecto al schwannoma melanocítico psamomatoso, una lesión neoplásica benigna derivada de las células de Schwann, se presenta en el 10% de los pacientes con CNC alrededor de los 20 años.
Con respecto a las lesiones hipofisarias, más de un 75% de los pacientes con CNC presentan aumento asintomático de la hormona de crecimiento (GH), del factor de crecimiento insulínico tipo 1 o de prolactina; sin embargo, en menos de 10% se evidencia tumor detectable. Es decir, las alteraciones bioquímicas pueden darse en ausencia de adenoma hipofisario35. Recientemente se publicó el único caso de corticotropinoma en un paciente con CNC36.
Genes relacionados con el complejo de CarneyGen PRKAR1ALas mutaciones inactivantes del PRKAR1A son responsables de las manifestaciones fenotípicas del CNC en más de un 70% de los casos5,6; estos son reconocidos como CNC tipo 1 (CNC 1).
Estas mutaciones en línea germinal también se han encontrado en casos aislados de PPNAD (OMIM 610489) sin expresión del resto de las manifestaciones del CNC31,32.
Hasta la fecha se han descrito más de 125 mutaciones diferentes del gen PRKAR1A en más de 400 familias de diversos orígenes étnicos con CNC (http://prkar1a.nichd.nih. gov/hmdb/intro.html).
La mayoría de las mutaciones dan como resultado la aparición de un codón de terminación. Son mutaciones «nonsense» que originan un ARN mensajero que codifica una proteína más corta de lo habitual (proteína truncada). Esta anomalía es detectada por un mecanismo de vigilancia de la transcripción genética o sistema de degradación del ARN mensajero mediada por mutación terminadora (NMD, por sus siglas en inglés). El mismo se inicia en el momento en que se detecta una codificación de proteínas truncadas que impedirían el funcionamiento celular normal, y da lugar a la degradación de este ARN; en consecuencia, no se produce la expresión de la proteína anómala detectable6,9,14,24,37. Por otra parte, si dichos cambios están hacia el final del gen, es muy posible que este sistema no se active, lo que da lugar a un fenotipo diferente en los pacientes con CNC, como se describirá más adelante38.
La mayoría de las mutaciones inactivantes del PRKAR1A originan una reducción del 50% de la expresión de la proteína RI-α, dado que únicamente se expresa el gen en el alelo no mutado. Este fenómeno, conocido como haploinsuficiencia del PRKAR1A, resulta de la incapacidad de un solo gen no mutado de mantener el fenotipo normal de un individuo, y se considera la base genética del CNC6,7,11,24,39.
Con menos frecuencia, también existen mutaciones del gen PRKAR1A que se expresan a nivel proteico por falta de activación de las NMD. Se trata de pequeñas inserciones y deleciones que no dan lugar a cambio en el marco de lectura y variantes tipo «splicing», que no llevan a la enfermedad por haploinsuficiencia proteica, sino por originar proteínas defectuosas que fallan en responder apropiadamente al AMPc38.
Sitios hotspotsA pesar de la heterogeneidad molecular del CNC se identificaron varios sitios calientes o «hotspots» del gen PRKAR1A donde se concentran la mayoría de las mutaciones relacionadas con la enfermedad. Por ejemplo, la mutación por cambio del marco de lectura («frameshift») c.491_492delTG (p.Val164Aspfs) en el exón 5, se encontró en más de 14 familias. Otros ejemplos son la deleción c.709-2_709-7delATTTTT hallada en 11 familias, y la variante de un solo nucleótido en el exón 2: c.82C>T (p.Gln28Ter) observada en varios pacientes con CNC5,6,24,39,40.
Correlación genotipo-fenotipoLos estudios multicéntricos han permitido establecer que ciertos grupos de pacientes con CNC presentan características clínicas del síndrome que se correlacionan con mutaciones específicas. Esta heterogeneidad genética de la enfermedad y la correlación con ciertos fenotipos clínicos refuerzan la importancia del estudio genético de los pacientes con CNC. Por ejemplo, la variante de mutación «hotspot» c.491_492delTG (p.Val164Aspfs) está asociada con la presencia de léntigos, mixomas cardiacos y nódulos tiroideos5,24,39,40.
Aquellos pacientes con PPNAD presentan con más frecuencia la mutación c.709-2_709-7delATTTTT y la sustitución c.1A>G que afecta el codón de iniciación de la proteína. Es difícil correlacionar la base molecular subyacente a esta expresión fenotípica, pero ambas originan un codón de terminación y activación del mecanismo de NMD5,6,9.
Por otra parte, las mutaciones del PRKAR1A que no están relacionadas con el mecanismo NMD, se asocian con una mayor expresión fenotípica de todas las manifestaciones de CNC5,6,9,38.
También se encontraron grandes deleciones del gen PRKAR1A (328bp a 3Mb) asociadas con una variedad de fenotipo más severo e inusual, posiblemente por un estado de haploinsuficiencia de genes adicionales40.
Por otro lado, en pacientes con criterios clínicos de CNC sin mutación del PRKAR1A se encontró un segundo locus del CNC; se trata de una región de 10Mb en el cromosoma 2p16 hallada por análisis de ligamiento. En tumores de pacientes con CNC se han encontrado cambios en el número de copias de esta región; este grupo se ha denominado CNC tipo 2 (OMIM 605244). Aunque el gen afectado en este sitio aún no ha sido identificado, dado que los fenotipos de CNC1 y CNC2 no son significativamente diferentes se ha indicado que los genes implicados podrían estar involucrados en la misma vía molecular6,12,24.
Fosfodiesterasa 11A partir de los hallazgos de cierta correlación entre genotipo y fenotipo en CNC, se postuló que la expresión de ciertos genes adicionales al PRKAR1A podría ser modificadora del fenotipo de la enfermedad. Este es el caso del gen que codifica a la fosfodiesterasa 11 (PDE11 por sus siglas en inglés). Esta es una enzima que tiene una acción dual: cataliza la hidrólisis del AMPc y del GMPc. Se expresa en varios órganos, y a nivel adrenal solo se expresa la variante splice A4 (PDE11A). Una disminución en la expresión de este gen da como resultado el aumento en los niveles del AMPc, de las proteínas fosforiladas por este y de toda la cascada inducida por el mismo. Esto sucede en el caso de mutaciones inactivantes del PDE11A, ubicado en el cromosoma 2q31_35, lo cual origina una proteína truncada41–47.
Por otra parte, también se describieron variantes genéticas del PDE11A con cambios en la secuencia del gen, que predisponen a la asociación entre tumores adrenales y testiculares en CNC. En algunos pacientes coexiste una mutación del gen PRKAR1A con estas diferentes variantes de PDE11A; en ellos se ha observado una asociación significativa entre desarrollo de PPNAD y tumores testiculares (tumor testicular de células grandes calcificadas de Sertoli), por lo que se considera un factor genético modificador del fenotipo42–47.
Gen de la subunidad catalítica del complejo proteína cinasa A (PRKACA)) y de la subunidad catalítica beta del complejo proteína cinasa A (PRKACB)Desde el año 2015, dentro de los criterios diagnósticos del CNC se incluyen los hallazgos de variantes activantes de los genes PRKACA y PRKACB9. Estos son diferentes del gen PRKAR1A en un principio relacionado con la enfermedad, pero que también están implicados en la vía del AMPc, antes descrita.
Con respecto al PRKACA que codifica a la subunidad catalítica C alfa (Cα) del complejo PKA, se encontró su sobreexpresión en diferentes fenotipos clínicos de enfermedad adrenal que afectan la síntesis de cortisol y que son causa de síndrome de Cushing ACTH independiente, aparte de la PPNAD clásicamente relacionada con el CNC.
La duplicación germinal del PRKACA se relaciona con hiperplasia adrenal bilateral, en sus diversas presentaciones, mientras que las mutaciones somáticas del mismo gen dan origen a adenomas suprarrenales unilaterales productores de cortisol9,30,31,48,49.
En pacientes con PPNAD como presentación aislada, se determinó la portación germinal de ganancia en el número de copias (duplicación) de la región genómica del cromosoma 19 que incluye al gen PRKACA. Por otra parte, la mutación somática c.617A>C (Leu206Arg) del mismo, se halló en pacientes con adenomas adrenales productores de cortisol30,31,48,49. Los estudios in vitro determinaron que esta mutación conduce a una activación constitutiva de Cα, con deterioro en la inhibición de la regulación de ambas subunidades catalíticas, ejercida en condiciones normales, por las subunidades reguladoras de la PKA. Además, se confirmó el aumento en los niveles proteicos de las subunidades catalíticas del complejo PKA en pacientes con ganancia en el número de copias del cromosoma 1930.
Por otra parte, con respecto al gen PRKACB, se encontró su sobreexpresión en una paciente con CNC que no tenía mutación del gen PRKAR1A ni alteración de PRKACA.
Se trata de una paciente de 19 años con manifestaciones de CNC que incluían acromegalia, lesiones pigmentadas cutáneas y mixomas con ausencia de síndrome de Cushing. El estudio genómico identificó una triplicación de 1,6Mb del cromosoma 1p31.1, que incluía el gen PRKACB, el cual codifica a la subunidad catalítica beta (Cβ), la segunda subunidad catalítica más importante de la PKA. El defecto fue confirmado por hibridación in situ, que mostró material genético adicional en el cromosoma marcado de forma supernumeraria. Los niveles de Cβ, pero no de Cα, se encontraron aumentados en linfocitos y fibroblastos de la paciente y en el material de un mixoma mamario. En sus linfocitos también se determinó actividad incrementada de la PKA en niveles similares a la de otros pacientes con CNC relacionados con mutaciones inactivantes del gen PRKAR1A50.
ConclusiónEl CNC es una rara entidad de herencia autosómica dominante, por lo que es importante el estudio genético de los casos índice y de sus familiares de primer grado.
Como en todas las enfermedades poco frecuentes, es importante desarrollar el seguimiento de los pacientes a nivel asistencial y la investigación de la base molecular mediante estudios multicéntricos, para que el aporte recíproco permita profundizar el conocimiento sobre las zonas oscuras de esta enfermedad.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.
