





El síndrome de Prader-Willi es un trastorno genético causado por alteraciones cromosómicas en el segmento 15q11-q13 que incluye sintomatología cognitiva, mental y conductual, así como un fenotipo somático específico. Tanto las alteraciones psicopatológicas más comunes (discapacidad intelectual, obsesiones, impulsividad, comportamientos de tipo autista, autolesiones) como las comorbilidades principales (cuadros afectivos, psicosis, trastorno obsesivo-compulsivo, trastorno del espectro autista) se caracterizan por una gran heterogeneidad, lo que justifica la necesidad de una mayor caracterización de su frecuencia y modo de presentación. Además de sus efectos sobre la composición corporal y la hipotonía, la hormona del crecimiento ha demostrado utilidad en el control conductual, así como algunos psicofármacos. También se han descrito alternativas a nivel experimental que están mostrando resultados alentadores. Un adecuado conocimiento de la psicopatología asociada a este síndrome permitiría mejorar el abordaje clínico, la identificación de los síntomas, la detección de comorbilidades y la instauración de un tratamiento más efectivo.
Prader-Willi syndrome is a genetic disorder caused by chromosomal changes in segment 15q11-q13 including cognitive, mental, and behavioral symptoms, as well as a specific physical phenotype. Both the most common psychopathological changes (intellectual disability, obsessions, impulsivity, autism spectrum disorders, self-injuries) and the main psychiatric comorbidities (affective disorders, psychosis, obsessive-compulsive disorder, autism spectrum disorder) are characterized by a great heterogeneity, which warrants the need for better identification of their frequency and clinical signs. In addition to its effects on body compositionand hypotony, growth hormone has been shown to be useful for regulating patient behavior, and psychoactive drugs are also an option. Other alternatives have shown promising results in experimental trials. Adequate understanding of the psychopathology associated to Prader-Willi syndrome would allow for improving clinical approach, symptom identification, detection of comorbidities, and administration of more effective treatments, leading to better clinical outcomes.
El síndrome de Prader-Willi (SPW) es un trastorno del neurodesarrollo de base genética, descrito por primera vez en 1956, que se caracteriza por un fenotipo físico y mental específico. Se le estima una prevalencia de entre 1:10.000 y 1:30.000 y está causado por una falta de expresión de los genes de origen paterno del segmento 15q11-q13. La alteración cromosómica responsable del trastorno condiciona el denominado «subtipo genético», que puede ser debido a microdeleción paterna (70-75% de los casos), disomía materna (20-25%), defecto de la impronta (1-3%) o translocaciones cromosómicas (menos del 1%)1.
Las manifestaciones físicas del SPW incluyen estatura baja, cifoescoliosis, hipopigmentación, hipoplasia genital debida a hipogonadismo hipogonadotropo, manos y pies pequeños, diámetro bifrontal estrecho, boca en forma de U invertida y anomalías oculares (miopía, estrabismo o fisuras palpebrales en forma de almendra). También se han objetivado disminución de los movimientos fetales durante el embarazo, hipotonía con dificultades para la succión en la etapa perinatal y letargia infantil, que mejora con la edad. Asimismo, resultan muy características la hiperfagia exagerada y la falta de saciedad, ya que determinan una ganancia de peso importante que se ha asociado a múltiples comorbilidades: hipertensión arterial, alteraciones del metabolismo lipídico, diabetes, apnea del sueño, etc. Estas complicaciones representan la principal causa de mortalidad en esta población2.
Aparte de las repercusiones somáticas, se han descrito igualmente síntomas mentales y conductuales asociados al SPW. El objetivo de esta revisión es actualizar su conocimiento, incidiendo en sus características clínicas y los diagnósticos psiquiátricos comórbidos. Posteriormente, se repasarán las particularidades en cuanto al tratamiento.
El cuidado integral de esta compleja enfermedad requiere de un equipo multidisciplinar que abarque no solo el ámbito sanitario, sino también los servicios sociales, comunitarios y educativos. Conocer las alteraciones psicopatológicas asociadas al SPW resultará imprescindible para el manejo clínico de los pacientes, ya que no siempre habrá disponibilidad de centros especializados en esta entidad. El endocrinólogo tendrá un papel crucial en este proceso, ya que en la mayoría de los casos será la principal consulta a la que acudan los pacientes una vez confirmado el diagnóstico.
Mecanismo fisiopatológicoEn los últimos años han proliferado los estudios que intentan hallar una base neuroanatómica a la psicopatología de los trastornos genéticos. Sin embargo, se dispone de poca bibliografía específica para el SPW.
La tomografía por emisión de positrones ha descubierto una hipoperfusión temporoparietal y del sistema límbico, así como alteraciones en el metabolismo glucídico de múltiples áreas cerebrales3. La resonancia magnética ha permitido cuantificar una disminución de la sustancia gris en el hipocampo y los lóbulos frontal y temporal; una disminución de la sustancia blanca en los lóbulos frontal y temporal, tronco encefálico y cerebelo y anomalías en la conexión de áreas corticales y ganglios basales, principalmente en relación con el hipotálamo4. Otros hallazgos característicos han sido la polimicrogiria en determinadas áreas, como en la cisura de Silvio, y la afectación del neurotransmisor GABA.
Todo ello ha postulado a las alteraciones en la migración neuronal y en la fase de replegamiento cerebral como posible mecanismo etiopatogénico. Estas variaciones estarían influidas a nivel cromosómico: se han descrito diferencias estructurales en función del subtipo genético (microdeleción paterna o disomía materna)5.
La consecuencia última de tales procesos es la disminución de la complejidad cortical, lo que la convertiría en causante (al menos parcial) de las alteraciones psicopatológicas que acompañan al SPW. Los déficits neuroendocrinológicos más asociados a la clínica somática (disfunción hipotalámica, hiposecreción de hormona del crecimiento y gonadotropinas…) también jugarían su papel, aunque su influencia a nivel mental todavía no ha sido evaluada a día de hoy.
Manifestaciones clínicasAl determinar diferencias estructurales, el subtipo genético también condiciona variaciones en las características clínicas del SPW. Este fenómeno ha sido ampliamente documentado en los 2subtipos genéticos principales (ver tabla 1) y se desarrollará de modo más extenso en cada uno de los apartados correspondientes.
Principales manifestaciones clínicas y comorbilidades del SPW en función del subtipo genético
| Predominio en microdelección paterna | Predominio en disomía materna | |
|---|---|---|
| Manifestaciones clínicas | Inestabilidad afectiva | Dificultades sociales |
| Peor rendimiento verbal | Impulsividad | |
| Conductas obsesivas | Rabietas, ira | |
| Autolesiones (rascados) | Oposicionismo, agresividad | |
| Obsesión por la comida | Conductas de tipo autista | |
| Comorbilidades | Trastorno obsesivo-compulsivo | Trastornos psicóticos |
| Trastorno del espectro autista |
Con relación a su evaluación clínica, únicamente existen 2escalas validadas de forma específica para el SPW. El Prader-Willi Syndrome Behavior Questionnaire (PSWBQ), desarrollado en 2014 por un grupo israelí, consta de 37 ítems que analizan las alteraciones de conducta de estos pacientes6. El Hyperphagia Questionnaire, algo más limitado, se centra exclusivamente en los problemas con la alimentación de la población con SPW7. Aunque no es específica para esta entidad, la Yale-Brown Obsessive Compulsive Scale también ha sido utilizada para la evaluación de los síntomas obsesivos.
A pesar de los intentos en esa dirección, no se dispone todavía de un modelo integral que permita explicar la psicopatología de este síndrome. El intento que se acerca más a este objetivo es probablemente el de Thuilleaux et al., que caracteriza a los pacientes con base en sus síntomas y define 4perfiles clínicos: básico, impulsivo, compulsivo y psicótico8. La revisión actual rehúye cualquier clasificación clínica y se limita a describir la psicopatología de manera global.
Discapacidad intelectual y otros déficits cognitivosLa capacidad cognitiva en el SPW oscila entre el rendimiento límite y la discapacidad intelectual moderada, con valores medios de CI de 65 puntos. Una revisión de la literatura puso de manifiesto que solo el 5% de los sujetos tiene un CI dentro de la normalidad9, lo que conduce frecuentemente a dificultades de aprendizaje y fracaso escolar.
En el ámbito neuropsicológico, los pacientes obtienen peores puntuaciones en funciones ejecutivas, reconocimiento facial y planificación y resolución de problemas. La memoria a corto plazo se afecta más que la memoria a largo plazo, mientras que los déficits en el procesamiento auditivo son más marcados que los déficits en el procesamiento visual10. Se han detectado puntuaciones «muy bajas» en integración visomotora y «por debajo de la media» en percepción visual y coordinación motora11.
La adquisición del lenguaje se presenta con retraso en comparación con la población sana: en algunos casos no llega hasta los 6 años de edad. Estas dificultades de comunicación se han descrito tanto en la emisión como en la recepción de la información, con resultados inferiores a lo esperable por su inteligencia verbal: aunque conservan la habilidad de definir palabras, se constatan limitaciones en la construcción de relaciones semánticas, en la comprensión oral y en la elaboración de frases.
Socialmente, al menos el 60% presenta problemas para identificar emociones, tolerar los cambios de rutina, jugar con chicos de su edad, comprender el concepto de distancia interpersonal y sentirse integrado. Comparados con población sana de entre 7 y 12 años, los niños con SPW muestran un retraso medio de 4 años en teoría de la mente (capacidad de comprender que los demás tienen sus pensamientos y de aceptar que pueden ser distintos a los propios)12. Todos estos déficits se traducen en una falta de habilidades sociales que causa progresiva tendencia al aislamiento y que, a diferencia de otros trastornos del neurodesarrollo, se agrava con la edad.
En cuanto al subtipo genético, los pacientes con microdeleción paterna puntúan mejor en habilidades sociales, mientras que los sujetos con disomía materna rinden mejor en el área verbal, pero son más lentos en el procesamiento cognitivo13.
Autolesiones e ingesta compulsivaDe forma asociada a los déficits cognitivos, el SPW se caracteriza por rigidez cognitiva, perseveración y dificultades para tolerar los cambios de rutina. En este contexto son muy habituales los comportamientos obsesivo-compulsivos, con un impacto significativo en el funcionamiento social y la calidad de vida. De todos ellos, destacan las autolesiones y la obsesión por la comida, a menudo ligada a una ingesta compulsiva; aunque menos asiduamente, se han descrito también la necesidad de orden y las conductas de acumulación (principalmente de alimentos). Tanto las autolesiones como la obsesión por comer se presentan con mayor frecuencia y gravedad en el subtipo microdeleción paterna14.
Hasta el 89% de los sujetos con SPW realiza actos autolesivos; los más frecuentes son los rascados cutáneos compulsivos (82%), que a menudo se asocian con heridas y sobreinfecciones. Otras conductas similares, aunque menos habituales, incluyen frotarse la nariz (28%) o el recto (6%), morderse las manos (17%), golpearse la cabeza (14%) o tirarse del pelo (9%)15. Según estudios transversales, los rascados cutáneos se incrementan durante la adolescencia y permanecen estables durante la edad adulta, con un cierto predominio en el género femenino16. Aunque se consideran generalmente como compulsiones, pueden verse influidos por la impulsividad del sujeto y se correlacionan con el grado de discapacidad intelectual. Estos comportamientos se verían incrementados en ausencia de supervisión de un adulto, lo que indica que podrían estar mantenidos por un refuerzo automático. Respecto a su etiopatogenia, se ha demostrado un aumento del umbral doloroso en estos pacientes al que podría estar contribuyendo una alteración en el procesamiento interoceptivo17.
En cuanto a la obsesión por comer, la resonancia magnética funcional la ha vinculado con el circuito de recompensa al detectar una disminución de las conexiones entre el núcleo estriado ventral y el sistema límbico18. Igualmente, se ha asociado a múltiples mecanismos neuroendocrinos implicados en la regulación del apetito19,20. En el supuesto de tener un acceso ilimitado al alimento, los pacientes con SPW consumirían el triple de calorías que los controles al aparearlos por IMC y edad: este impulso por la ingesta es tan intenso que algunos autores lo han equiparado a la dependencia de tóxicos, al definirlo como una verdadera «adicción a la comida»21,22. Esta obsesión, ligada a la impulsividad que presentan, condiciona compulsiones de búsqueda de alimento que se han relacionado con una mayor mortalidad por accidentes, sobre todo en los varones jóvenes23, además de las comorbilidades físicas ya mencionadas. En algún caso más extremo, han llegado a aparecer complicaciones del orden de la potomanía o incluso la pica. No se ha visto que las alteraciones conductuales asociadas a la comida disminuyan con la edad en ninguno de los 2subtipos genéticos.
Otras alteraciones del comportamientoLos pacientes con SPW suelen mostrar fluctuaciones del estado de ánimo e incapacidad para controlar sus emociones, lo que deriva en rabietas, impulsividad e incluso agresividad física en el 83-97% de los casos24. Al compararlas con las de niños sanos, estas conductas violentas tienen un inicio más tardío, una mayor severidad y una mayor persistencia en el tiempo, hasta que empiezan a disminuir a partir de los 19 años25. Los arranques de ira parecen ser la expresión de frustraciones, pero también pueden atribuirse a sentimientos de inferioridad, falta de empatía e incapacidad para entender las motivaciones de los demás. Es en este marco en el que se debe considerar el oposicionismo y la hostilidad que a menudo acompañan al SPW, las mentiras, los pequeños robos y otras actitudes posesivas o manipulativas. Se ha detectado una mayor prevalencia de estas alteraciones en pacientes con disomía materna26.
Finalmente, cabe mencionar también algunos comportamientos repetitivos que recuerdan a los de pacientes autistas, como los tics y las conductas estereotipadas. Los intereses restringidos (típicamente los puzles) y las dificultades en la interacción social (ya comentadas con anterioridad) serían otras manifestaciones compartidas con el autismo. Estas actitudes aberrantes también predominan en el subtipo disomía materna27 y pueden aparecer en ausencia o en presencia de un trastorno del espectro autista (TEA) comórbido. El estudio genético, los síntomas acompañantes y el fenotipo físico contribuirán al diagnóstico diferencial.
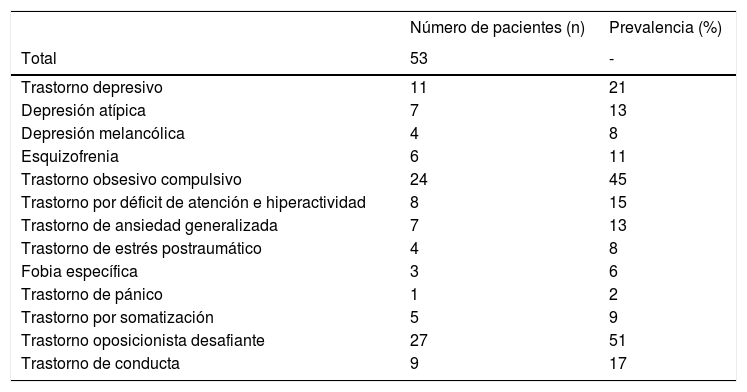
ComorbilidadesLa asociación del SPW con otros diagnósticos psiquiátricos viene condicionada por multitud de factores genéticos, biológicos y ambientales, y se ha detectado hasta en el 89% de los casos (ver tabla 2)28. Su presentación suele ser atípica en las fases más iniciales, por lo que cualquier cambio en los patrones de comportamiento habitual debería ser sugestivo de comorbilidad.
Prevalencia de las principales comorbilidades psiquiátricas en una población de 53 pacientes (adolescentes y adultos) con diagnóstico de SPW
| Número de pacientes (n) | Prevalencia (%) | |
|---|---|---|
| Total | 53 | - |
| Trastorno depresivo | 11 | 21 |
| Depresión atípica | 7 | 13 |
| Depresión melancólica | 4 | 8 |
| Esquizofrenia | 6 | 11 |
| Trastorno obsesivo compulsivo | 24 | 45 |
| Trastorno por déficit de atención e hiperactividad | 8 | 15 |
| Trastorno de ansiedad generalizada | 7 | 13 |
| Trastorno de estrés postraumático | 4 | 8 |
| Fobia específica | 3 | 6 |
| Trastorno de pánico | 1 | 2 |
| Trastorno por somatización | 5 | 9 |
| Trastorno oposicionista desafiante | 27 | 51 |
| Trastorno de conducta | 9 | 17 |
Fuente: Adaptado de Shriki-Tal et al.28.
De forma general, el 10-15% de los individuos con SPW desarrollará síntomas psicóticos al final de la adolescencia o al inicio de la edad adulta. En la mayoría de los casos, estos síntomas asociarán un componente afectivo y se presentarán a menudo como fases hipomaníacas o depresivas. El riesgo de padecer un trastorno afectivo se ha cifrado en un 17,4%; por el contrario, no se ha logrado confirmar una mayor prevalencia de esquizofrenia, a pesar de existir alteraciones genéticas y epigenéticas comunes29.
Existen diferencias de prevalencia según el subtipo genético de los pacientes. Con relación a los síntomas psicóticos, el riesgo es muy elevado (60%) en aquellos casos con disomía materna y disminuye hasta el 20% con microdeleción paterna30. La presentación clínica suele ser en forma de psicosis cicloides que asocian conductas paranoides, alucinaciones auditivas y cambios de humor de inicio subagudo e intensidad variable; aunque también se han descrito patrones episódicos de euforia o depresión más compatibles con un trastorno bipolar.
Además de padecer estos síntomas con más asiduidad, los sujetos con disomía materna suelen tener un pronóstico más desfavorable, un mayor riesgo de recurrencia psicótica y, posiblemente, una peor respuesta al tratamiento31. Este fenómeno no se produce en las recaídas depresivas, que se presentan con una prevalencia del 20-25% en ambos subtipos genéticos32. La microdeleción paterna, en cambio, se ha vinculado en los varones con un mayor riesgo de depresión, agresividad y trastorno dependiente de personalidad33. A pesar de ello, generalmente se considera que, obtenida la estabilidad con tratamiento antipsicótico, las recurrencias son más bien infrecuentes.
La catatonía puede ser otra de las manifestaciones de la psicosis, como se ha descrito en series de casos. A pesar de ello, no disponemos de bibliografía específica acerca de su prevalencia, particularidades clínicas o abordaje terapéutico en la comorbilidad con el SPW.
Respecto a su etiología, el desarrollo de los síntomas psicóticos podría estar relacionado con una sobreexpresión del material genético materno. Un estudio de 201734 indica que el alto riesgo de psicosis en individuos con disomía materna puede resultar de un desarrollo microestructural aberrante de la sustancia blanca. En cuanto a los síntomas afectivos, el pequeño fragmento de ARN nucleolar (sno)ARN HBIII-52 ha sido implicado al alterar el receptor 2C de serotonina35. Aparte de estos hallazgos, el mecanismo causal responsable sigue siendo una incógnita. Los esfuerzos actuales deberían dirigirse a la identificación de aquellos pacientes con SPW que presentan mayor riesgo de desarrollar psicosis, lo que permitiría caracterizar mejor los síntomas prodrómicos y ayudaría al diseño de ensayos clínicos.
Trastorno del espectro autistaAunque la mayoría de los individuos con SPW no cumple estrictamente criterios de TEA, existen semejanzas en su presentación clínica: la rigidez cognitiva, la perseveración, la reticencia al cambio, los rituales, los comportamientos repetitivos, los intereses restringidos y las dificultades sociales son rasgos compartidos entre ambos diagnósticos. Algunas de las mutaciones identificadas en el segmento 15q11-q13 están presentes tanto en el SPW como en el TEA, lo que podría indicar un trasfondo genético común.
Según la evidencia disponible, esta comorbilidad es habitual, aunque también está influida por el subtipo genético: en pacientes con TEA idiopático, la disomía materna se presenta en el 1-3% de los casos, mientras que en la microdeleción paterna lo hace con menos frecuencia36. Estos datos, basados en entrevistas familiares y escalas clínicas, explican la mayor coincidencia de ambos diagnósticos en el subtipo disomía materna (35-37,7%), con valores muy inferiores en el caso de la microdeleción paterna (18-18,5%)37.
Además del subtipo genético, se ha conseguido correlacionar la presencia de TEA con el género masculino, pero no con el tratamiento farmacológico, el IMC, el estatus socioeconómico o el nivel educativo de los padres. A grandes rasgos, los sujetos con la comorbilidad, a diferencia de aquellos con SPW sin TEA, presentan más estereotipias e intereses restringidos y puntúan peor en CI y en habilidades sociales. Las autolesiones más comunes en el SPW son los rascados cutáneos, mientras que en el TEA predominan las mordeduras o los golpes. En comparación con el síndrome de Asperger, se ha descrito una temporalidad diferente en el desarrollo de las conductas de tipo autista, que serían poco prevalentes en la primera infancia, más prominentes a partir de los 6 años y equiparables a las del síndrome de Asperger a partir de los 13 años de edad38.
Trastorno obsesivo-compulsivoNo queda claro hasta qué punto los comportamientos obsesivo-compulsivos típicamente asociados al SPW (obsesión por la comida, rascados compulsivos…) podrían solaparse con un trastorno obsesivo compulsivo (TOC) bien instaurado. En efecto, algunas de las alteraciones en la conectividad cerebral descritas para el TOC (tanto entre estructuras subcorticales como entre el córtex prefrontal y los ganglios basales) pueden hallarse también en los pacientes con SPW. Como ya se ha visto, los síntomas obsesivos se presentan con mayor frecuencia y severidad en los pacientes con microdeleción paterna, lo que haría esperable una mayor prevalencia de TOC comórbido en esta subpoblación, que se ha cifrado en un 26%39.
En cuanto al diagnóstico diferencial, los pensamientos obsesivos en el SPW son menos aparentes que las compulsiones. Las compulsiones predominantes guardan más relación con las autolesiones y la búsqueda de comida, mientras que aquellas clásicamente asociadas al TOC (lavado de manos, comprobaciones) se presentan más raramente. De forma característica, estos actos compulsivos no responden a la necesidad de aliviar la ansiedad o el estrés, sino que simplemente se realizan porque resultan agradables40.
Trastorno por déficit de atención e hiperactividadLas alteraciones conductuales en el SPW incluyen la labilidad emocional, las rabietas y los arranques de ira, que son comunes con el trastorno por déficit de atención e hiperactividad (TDAH). La evidencia disponible sobre la coincidencia de ambos diagnósticos es más bien escasa. Según un estudio de 200541, la prevalencia de TDAH alcanzó el 25,9% en una cohorte de 58 pacientes con SPW; de modo clínicamente significativo, también se registraron problemas de conducta en el 22,4% de los casos y problemas de impulsividad e hiperactividad en el 19%. Otro estudio de características similares42, realizado en 2012 en 24 pacientes con SPW, mostró una prevalencia de TDAH del 21%. Cabe señalar que estos resultados están basados en escalas administradas a familiares, lo que podría constituir un sesgo, al no haberse realizado una valoración directa de los pacientes.
Trastornos del sueñoDebido a la hiperfagia y la obesidad, resulta muy habitual el síndrome de apnea obstructiva del sueño, presente en el 79,9% de los casos43. Por lo general, se recomienda su cribado sistemático y, en caso de sospecha, la derivación a un neumólogo especialista.
La somnolencia diurna excesiva acompaña con frecuencia al SPW, tiene su inicio en la infancia y se presenta con mayor frecuencia que en otras discapacidades intelectuales. No es infrecuente que, durante los primeros años de vida, los sujetos se muestren somnolientos a lo largo del día y, ya mayores, refieran síntomas de tipo cataplejía hasta en el 16% de los casos (aunque, por norma general, no suele haber otra clínica sugestiva de narcolepsia)44. El gen SNORD116, que se altera en el SPW, ha sido implicado tanto en el control de los ritmos circadianos como en el de la hiperfagia45; de hecho, la somnolencia diurna excesiva persiste a pesar del tratamiento del síndrome de apnea obstructiva del sueño y se ha correlacionado positivamente con las alteraciones de conducta que acompañan al SPW.
Otras alteraciones comunes son los ronquidos, las dificultades para despertar y la disminución de la latencia del sueño REM.
Otras comorbilidadesEl aumento en los niveles de ansiedad y los sentimientos de inseguridad se traducen a menudo en hipersensibilidad al estrés, incapacidad para tolerar la incertidumbre, quejas somáticas, dependencia y necesidad constante de ser tranquilizados. Un estudio descriptivo sobre población adulta46 registró una mayor prevalencia de trastorno de ansiedad y de trastorno adaptativo de predominio ansioso (38%). En la misma muestra se hallaron también casos de trastorno explosivo intermitente (30%) con un predominio 2:1 de varones. Otro estudio realizado en pacientes más jóvenes (de entre 7 y 17 años de edad)47 desveló una frecuencia aumentada de trastorno oposicionista desafiante (20%) con independencia de la edad, el género, el subtipo genético o el CI.
TratamientoEl uso de fármacos en el SPW ha mostrado un efecto limitado en el control conductual de los pacientes. Sin embargo, su administración es común tanto para el manejo de los síntomas como en el caso de comorbilidades.
El único tratamiento aprobado de forma específica en niños con SPW es la hormona del crecimiento, que ha demostrado reducir la hipotonía y el riesgo de obesidad, normalizar parcialmente el desarrollo físico y psicológico y mejorar la capacidad cognitiva, disminuyendo los problemas de conducta48. Otros estudios señalan también una mejoría de la clínica depresiva y las capacidades hedónicas, aunque a riesgo de potenciar los síntomas TDAH49. En adultos, más allá de sus beneficios metabólicos, se ha probado su utilidad en la mejoría de la agilidad mental, la flexibilidad cognitiva, el tiempo de reacción, la capacidad atencional y la calidad de vida. No obstante, según la mayoría de las guías clínicas, hace falta demostrar un déficit de hormona del crecimiento, con tests de estimulación, para su indicación en población adulta, lo que sin duda dificulta su uso. Su administración es controvertida por la posibilidad de efectos adversos, pero solventable con un buen seguimiento50.
La combinación de naltrexona y bupropion tiene un efecto sinérgico en la población general que permite reducir la ingesta y la ganancia de peso. Por este motivo, ha sido utilizada en el SPW para el tratamiento de la hiperfagia y la impulsividad, aunque no se dispone de ensayos clínicos al respecto51. De forma preliminar, el uso de metformina en niños con SPW que presentan resistencia a la insulina e intolerancia a la glucosa también habría conseguido mejorar los cuestionarios de hiperfagia, la angustia relacionada con los alimentos y la capacidad para permanecer alejados de la comida52. A nivel experimental, se han obtenido buenos resultados en el control alimentario con la administración de análogos de GLP1 (exenatide53, liraglutide54) y de un análogo de la ghrelina desacilada55, pero no con análogos de somatostatina de larga duración (que, a pesar de modificar los niveles de ghrelina, no lograron cambios en el peso, la composición corporal, el apetito o la actitud hacia la comida)56. Por el momento, el resto de las alternativas farmacológicas no han demostrado ser eficaces en la reducción de la ingesta; la cirugía bariátrica, al no disminuir la hiperfagia ni conseguir pérdida de peso mantenida, tampoco es útil a medio/largo plazo57.
Considerando la alta frecuencia de alteraciones conductuales, la mayoría de los clínicos opta por utilizar antipsicóticos a dosis bajas. De todos ellos, la risperidona sería el fármaco más extendido58, aunque también se ha descrito buena respuesta a aripiprazol, quetiapina, clorpromazina, haloperidol y ziprasidona. Un estudio de 2015 puso de manifiesto que, de forma paradójica, el uso de antipsicóticos en el SPW podría asociarse a pérdida de peso59, probablemente al conseguir un mejor control conductual. El tratamiento sustitutivo para el hipogonadismo, presente hasta en el 50% de los casos, se ha demostrado seguro y no implica necesariamente un aumento de la agresividad ni de los problemas de conducta60,61.
En cuanto a los síntomas obsesivos, los inhibidores selectivos de la recaptación de serotonina constituyen la alternativa gold standard para el control de las rabietas y las compulsiones. De todos ellos, la fluoxetina sería el antidepresivo con más experiencia en este campo58, aunque no existe evidencia de su utilidad como inhibidor del apetito en pacientes con SPW. El topiramato sería otra opción igualmente aplicable en la reducción de las autolesiones y la agresividad por su efecto antiimpulsivo; su capacidad para disminuir las conductas relacionadas con la comida, no obstante, ha obtenido resultados contradictorios62. Las benzodiacepinas se han utilizado con éxito como tratamiento de soporte, pero se recomienda evitarlas por el riesgo de iatrogenia.
Más allá de los fármacos, resulta indispensable un abordaje ambiental, lo que incluye a menudo la necesidad de mantener la comida bajo llave, la supervisión rigurosa de la ingesta, dieta y ejercicio y el asesoramiento nutricional especializado. Las medidas conductuales (acompañamiento, mantener las uñas cortas) y los mecanismos de barrera (vendajes, aplicación tópica de antibióticos) reducirían también las autolesiones cutáneas y la posibilidad de infecciones. Psicoterapias como el análisis conductual aplicado, de utilización en el trastorno autista, han mostrado igualmente resultados prometedores en el manejo de las alteraciones comportamentales. Se debe resaltar la importancia de facilitar la transición de Pediatría a Adultos y de evitar la pérdida de seguimiento de estos pacientes por parte de Endocrinología63.
Existen muy pocos estudios que analicen el tratamiento de las comorbilidades psiquiátricas del SPW; en consecuencia, la concurrencia de otro trastorno mental se abordará de forma empírica con las alternativas convencionales, sea con medicación o con psicoterapia. El uso de antipsicóticos, independientemente del subtipo genético y de la historia de episodios psicóticos previos, ha demostrado disminuir el riesgo de trastorno psiquiátrico mayor durante el seguimiento64.
Hallar nuevas estrategias para el manejo de estos pacientes se ha convertido en un reto para la comunidad científica actual. La administración de oxitocina podría asociarse a un mejor rendimiento social y conductual, aunque los resultados por el momento no son concluyentes65. Otra alternativa prometedora sería la N-acetilcisteína, un modulador de la vía excitatoria glutamatérgica, que se ha relacionado con una reducción de los rascados cutáneos66. En un entorno controlado, la estimulación transcraneal de corriente directa67 y la terapia de estimulación del nervio vago68 podrían disminuir los comportamientos aberrantes asociados a la comida. La somnolencia diurna excesiva, de forma específica, ha presentado mejoría con modafinil en algunos casos69.
Resulta altamente recomendable la lectura de la Guía de actuación en el síndrome de Prader-Willi, elaborada por el sistema público de salud del País Vasco y dirigida a profesionales sanitarios, que desarrolla varias de las tesis expuestas en este artículo de forma más amplia70. Lo mismo ocurre con el capítulo 7 de la monografía Enfermedades de la impronta: guías de buena práctica clínica, que ha sido redactada por miembros de nuestro equipo y ofrece una revisión integral de este trastorno genético y sus opciones terapéuticas71. En cuanto a información para usuarios y sus cuidadores, el Manual para familiares de personas afectadas por el síndrome de Prader Willi, editado por la Asociación Síndrome Prader-Willi de Andalucía (ASPWA), recoge experiencias y testimonios de los propios afectados y de sus familiares y los transmite en un lenguaje asequible y apto para todos los públicos72.
ConclusionesEl SPW es un trastorno genético que se caracteriza por un fenotipo físico y mental específico. Las alteraciones psicopatológicas asociadas incluyen déficits cognitivos, discapacidad intelectual, dificultades en la interacción social, obsesión por la comida, rascados compulsivos, rabietas, fluctuaciones del ánimo, impulsividad y conductas de tipo autista. La comorbilidad con otros trastornos psiquiátricos es un hallazgo habitual que a menudo dificulta el diagnóstico y condiciona un abordaje integral.
El manejo del SPW requiere de una intervención biopsicosocial para el control óptimo de los síntomas. Los programas rehabilitadores de tipo multidisciplinar son la herramienta esencial para el tratamiento de los pacientes y el apoyo de sus familiares y cuidadores. De modo general, se suele optar por una combinación de psicoterapia, psicofármacos e intervenciones específicas para el control del comportamiento. Al no disponer de guías clínicas basadas en la evidencia, se recomienda individualizar el tratamiento en función de las necesidades de cada caso.
Conflicto de interesesLos autores declaran la ausencia de conflictos de interés.
