




El angioedema hereditario (AH) es un proceso infrecuente, de carácter recurrente, potencialmente mortal, originado por el déficit o disfunción de factor C1 inhibidor. El dolor abdominal secundario a edema intestinal es relativamente frecuente en pacientes con AH pero tras una revisión de la literatura solo se han informado seis casos de pancreatitis aguda asociado a angioedema hereditario.
Hereditary angioedema (HAE) is an infrequent, recurrent, and potentially lethal disorder caused by a deficiency of C1 inhibitor or its activity. Abdominal pain secondary to bowel edema is common in these patients. However, a thorough literature search yielded only six previously reported cases of pancreatitis associated with this entity.
El angioedema hereditario (AH) es una enfermedad infrecuente, que se transmite con herencia autonómica dominante, caracterizada por episodios recurrentes de angioedema de piel y mucosas a nivel de tubo digestivo y vías respiratorias, con resolución espontánea en menos de 48h, debido a un déficit del factor C1 inhibidor o a una disminución de la actividad del mismo. En un 25% de los casos se detecta una mutación espontánea1. La afectación gastrointestinal más frecuentemente asociada a AH son los episodios de dolor abdominal agudo recurrentes, asociados a nauseas y vómitos, simulando cuadros de abdomen agudo, que en ocasiones llevan a la realización de laparotomías «en blanco»2. La aparición de pancreatitis aguda (PA) es un hecho excepcional, habiéndose informado solo seis casos previamente.
Observación clínicaPaciente de 49 años, varón, con antecedentes personales de amigdalectomía en la infancia, ex-fumador de 20paq/año desde hacía diez años, hipertensión arterial sistémica, hipercolesterolemia y AH tipo 2. Seguía tratamiento con sinvastatina 20mg/d y enalapril 10mg/d. No tenía antecedentes personales de alcoholismo, enfermedad biliar o fibrosis quística, ni antecedentes familiares de enfermedad pancreática. El paciente había sido diagnosticado de AH diez años antes, tras haber presentado un cuadro de angioedema facial y de miembros superiores, con resolución «ad integrum» en 24h y haberse objetivado una disminución de la actividad del factor inhibidor de C1 (IC1). El paciente había rechazado tratamiento con danazol por sus efectos secundarios y presentaba uno o dos episodios de edemas de predominio en miembros superiores, de duración inferior a 48h, mensualmente. El paciente había sido estudiado en diferentes centros por cuadro de dolor abdominal crónico recurrente de años de evolución de causa no aclarada tras estudios radiológicos y endoscópicos. Tras detectarse helicobacter pilory, se indicó triple terapia (claritromicina, amoxicilina y omeprazol) durante 10 días y se confirmó posteriormente la erradicación del mismo mediante test de aliento.
Tres meses antes del ingreso actual, el paciente había estado hospitalizado durante cuatro días por episodio de dolor abdominal y ascitis ecográfica leve con resolución espontánea a las 48h del cuadro, con normalidad de las pruebas analíticas y sin detección de líquido libre intraabdominal al alta.
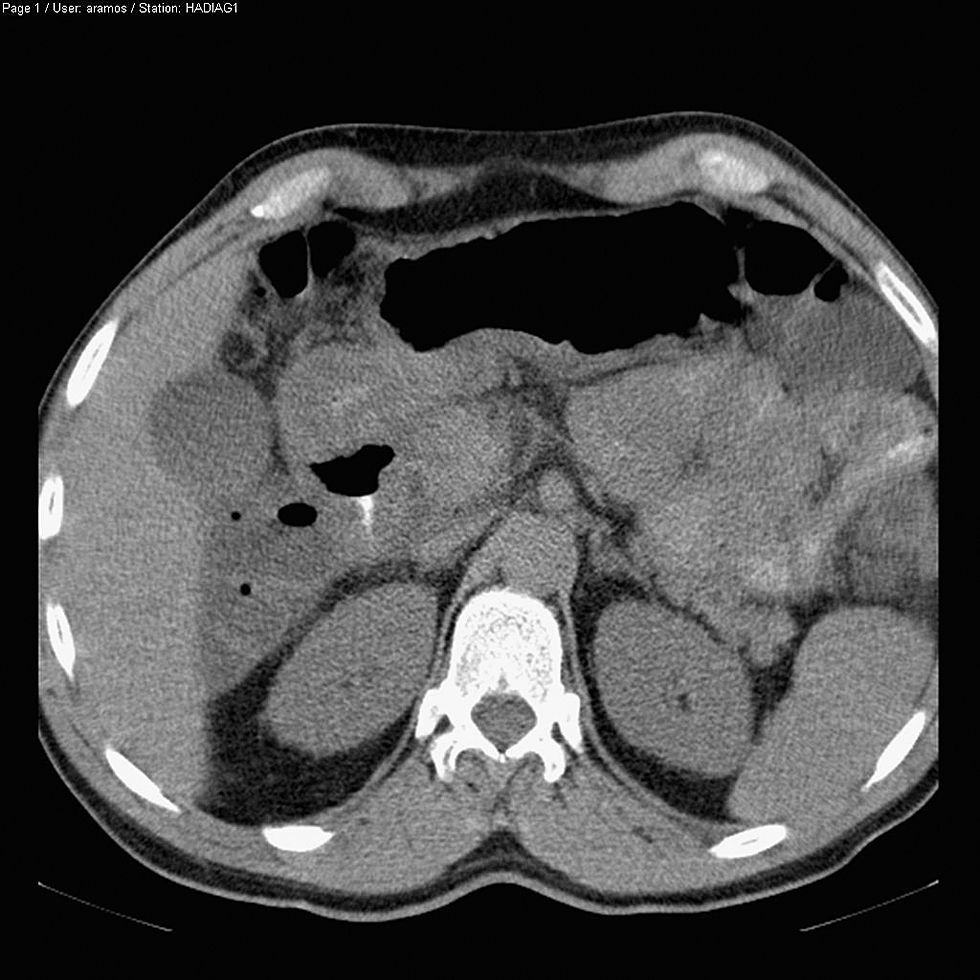
El paciente acudió a urgencias de nuestro centro hospitalario por un cuadro de dolor epigástrico de comienzo brusco, intenso, e irradiado hacia la espalda, de 6h de evolución, acompañado de nauseas, vómitos y despeños diarreicos. A la exploración física destacaba sensación de enfermedad, con signos mucocutáneos de deshidratación. No presentaba signos de inestabilidad hemodinámica. La auscultación cardiorrespiratoria era normal. El abdomen era blando y depresible, con dolor a nivel del epigastrio, sin signos de irritación peritoneal y ruidos hidroaéreos ligeramente disminuidos. Asimismo, se objetivó tumefacción indolora en ambas manos y antebrazos. En la analítica de urgencias destacaba: leucocitosis (Leu: 27.800×109/l), hematocrito: 53% creatinina: 2,15mg/dl, lipasa: 2.342U/l (valores normales: 114–286), amilasa: 445U/l (valores normales: 25–115). El paciente fue sometido a una tomografia axial computarizada (TAC) abdominal urgente por sospecha de perforación de víscera hueca, informándose de un páncreas ligeramente aumentado de tamaño, pero sin exudados peripancreáticos ni colecciones intraabdominales (fig. 1), si bien el estudio se realizó sin contraste intravenoso por la disfunción renal. El paciente permaneció en la unidad de cuidados intensivos durante 48h por insuficiencia renal aguda que se normalizó tras fluidoterapia bajo control de presión venosa central. La evolución clínica del paciente posteriormente fue adecuada, con desaparición del dolor abdominal y del angioedema cutáneo, con buena tolerancia oral y sin desarrollo de complicaciones, siendo dado de alta tras nueve días de ingreso hospitalario. Se solicitaron niveles de C4: 3,7mg/dl (valores normales: 16–38mg/dl) y de actividad de C1 inhibidor: <10% (valores normales: 70–130%).

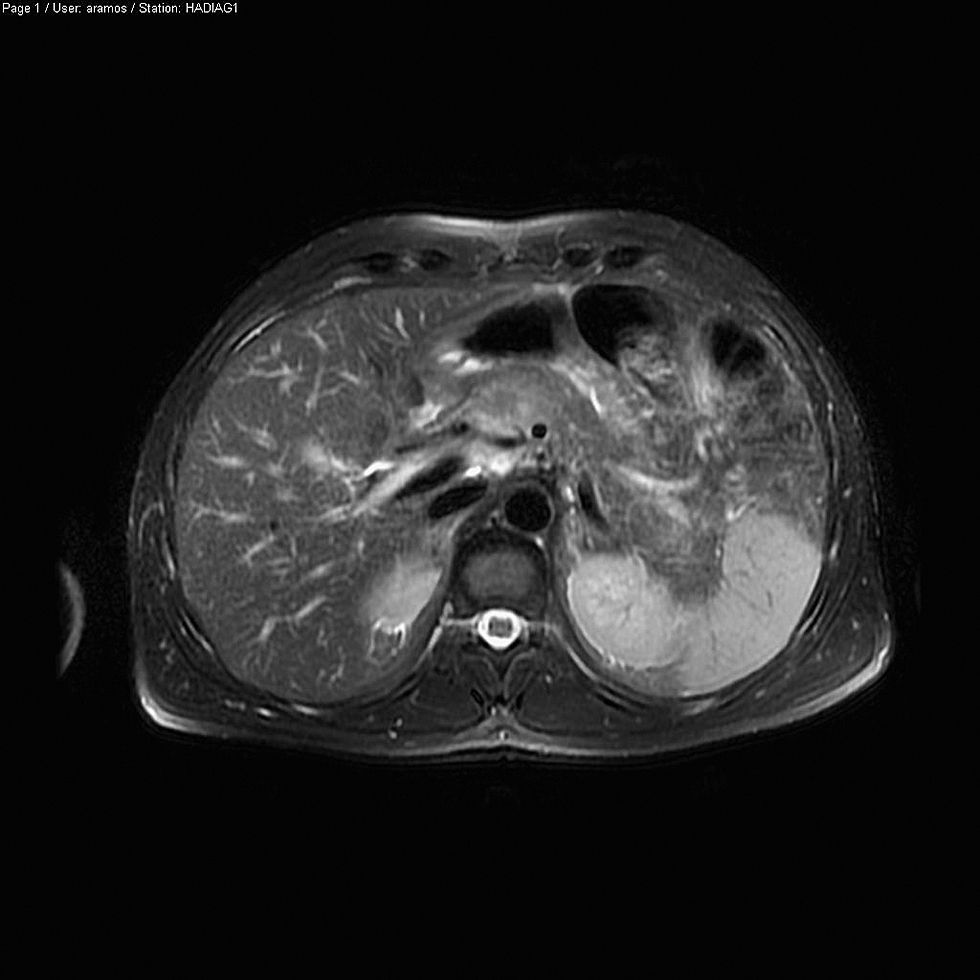
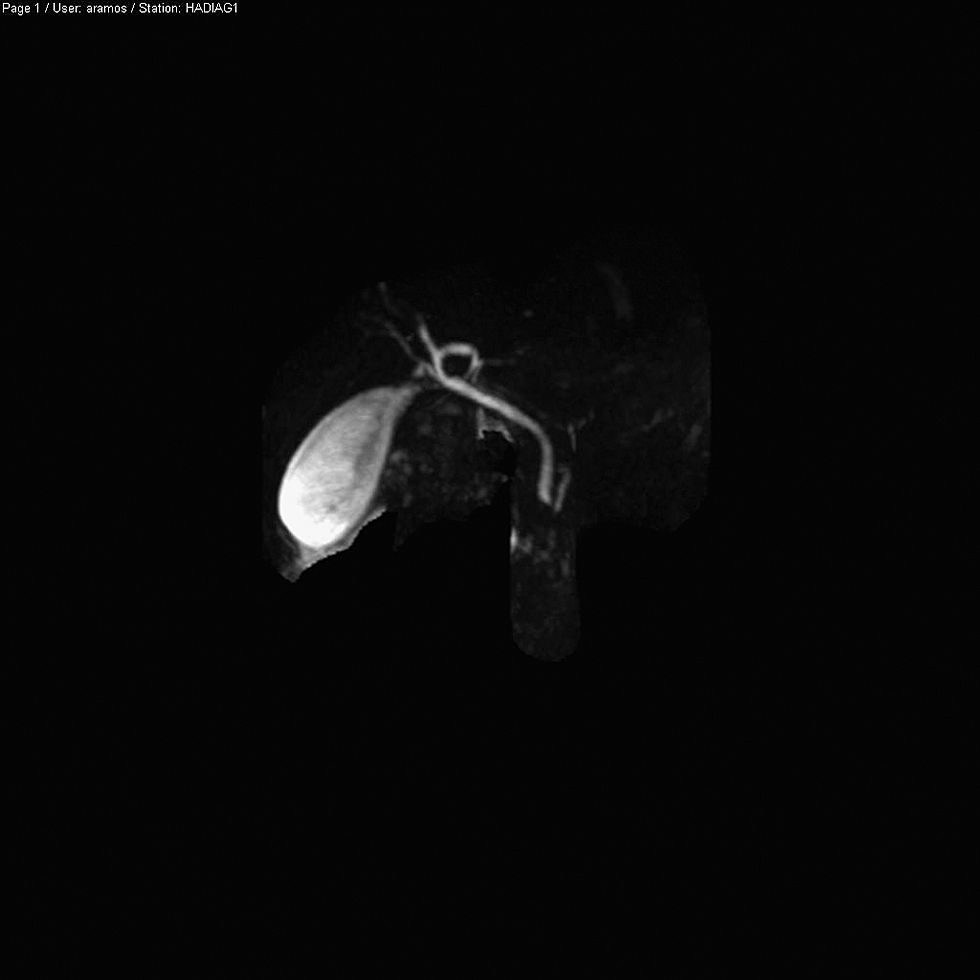
Los niveles de calcio, paratohormona, triglicéridos e inmunoglobulina G4 fueron normales. Se solicitó durante el ingreso una resonancia nuclear magnética de vías biliares y páncreas en la que se apreciaba un páncreas con aumento de la intensidad de señal, sin detectarse signos radiológicos de litiasis, anomalías en la vía biliar o en el conducto de Wirsung, siendo compatible por tanto con PA leve (figs. 2 y 3). Posteriormente se realizó una ecoendoscopia digestiva que no detectó la presencia de microlitiasis ni anomalías estructurales ecográficas a nivel de páncreas ni de la encrucijada biliopancreática. Al alta se le prescribió 20mg diarios de ácido tranexámico y posteriormente le fue suspendido el enalapril, a los tres meses del alta, por riesgo de recidiva de crisis de angioedema. Durante doce meses de seguimiento el paciente no ha vuelto a presentar nuevas crisis de dolor abdominal ni angioedema cutáneo.
Discusión

El AH es una enfermedad infrecuente que se caracteriza por episodios recurrentes de edema sin urticaria a nivel de tejido subcutáneo o submucoso en distintas localizaciones, que en algunas ocasiones incluye la laringe, originando la muerte en el 20% de estos casos1. A nivel gastrointestinal el edema submucoso puede originar dolor abdominal intenso, vómitos, diarrea y estreñimiento2, siendo extremadamente infrecuente la aparición de PA, de hecho, solo se han informado en nuestro conocimiento de seis casos previamente3–7.
Las primeras descripciones de la enfermedad datan de la primera mitad del siglo xix pero su carácter hereditario, en cinco generaciones consecutivas de una misma familia, fue descrita por primera vez en 1888 por Osler8. Se trata de una enfermedad con marcada tendencia familiar que se transmite mediante herencia autosómica dominante (AD), con penetrancia incompleta. Se han descrito más de cien mutaciones asociadas al gen del C1 inhibidor (C1H), el cual está situado en el cromosoma 11 (región q12–q13.1), siendo la expresión fenotípica de este gen muy variable9. Se ha informado una tasa de mutaciones espontáneas en torno al 25%. Se desconoce la prevalencia real del AH pero se estima en torno a 1:10.000 a 1:150.000 casos, sin diferencias significativas en cuanto a raza o sexo10.
La inhibición de C1 (IC1) tiene actividad reguladora sobre el sistema del complemento, la calicreína y el factor xii de la coagulación, por lo que su déficit origina una aumento de la liberación de la bradicinina y cininas del complemento, que conducen a una mayor permeabilidad vascular y secundariamente a una extravasación de liquido al espacio intersticial10,11.
Se distinguen tres tipos de AH:
- 1.
AH tipo i: representa el 85% de los casos. Se demuestra un déficit de la cantidad de proteína IC1 en plasma, en torno al 5–30% de las cifras normales.
- 2.
AH tipo ii: representa el 15% de los casos, en los que los niveles plasmáticos de la proteína IC1 son normales pero su funcionamiento se encuentra alterado.
- 3.
AH tipo iii: descrita recientemente en mujeres con clínica típica pero con una concentración y función normales de IC1 y un C4 normal12.
La edad de comienzo de los síntomas es variable, pero hasta el 40% de los casos los manifiestan en los primeros cuatro de años de vida. La sintomatología suele empeorar durante la adolescencia y persistir a lo largo de toda la vida. En los pacientes no tratados la frecuencia de repetición de los episodios, oscila entre siete y catorce días, si bien el curso de enfermedad es impredecible. Entre los factores desencadenantes se describen: traumatismos, menstruación, embarazo, infección por Helicobacter pylori, estrógenos, fármacos… pero con frecuencia no se detecta claramente el factor precipitante13–15. La incidencia de enfermedades autoinmunes está incrementada tales como las glomerulonefritis, el lupus eritematoso sistémico, la tiroditis autoinmune y las enfermedades inflamatorias intestinales16.
El síntoma guía es la aparición de episodios de edema no doloroso ni pruriginoso, de localización usualmente en manos y pies, pero que también puede afectar a genitales, cara, lengua y laringe, resolviéndose espontáneamente en 48–96h. Una tercera parte de los casos desarrollarán eritema marginado en el tronco o extremidades como pródromos1.
Nuestro paciente había sido diagnosticado diez años antes de AH tipo 2 y dado que el paciente no había presentado ninguna crisis de angioedema grave, asociado a que su frecuencia de episodios era baja, además de los posibles efectos secundarios del danazol, se optó por no prescribir tratamiento profiláctico. Tres meses antes del ingreso actual por la PA, el paciente había sido hospitalizado por un cuadro de dolor abdominal agudo, periumbilical, de características cólicas, sin alteraciones analíticas significativas pero en el que se había visualizado en la ecografía abdominal de urgencias una mínima cantidad de líquido libre intraabdominal, que posteriormente a las 48h no se objetivó, por lo que es probable que se tratara de un cuadro de angioedema abdominal dada su rápida y favorable evolución.
Las crisis de dolor abdominal en pacientes con AH es un evento muy frecuente; se ha informado que hasta el 97% de los pacientes afectos de AH las experimentan a lo largo de su vida y que hasta el 50% de las crisis de angioedema son abdominales17. El dolor abdominal suele ser de características cólicas, y se acompaña de nauseas y vómitos. En una serie retrospectiva, publicada en 2006, de 153 pacientes afectos de AH que habían en conjunto presentado 33.617 crisis de dolor abdominal, se valoró la intensidad del dolor abdominal, en una escala del 1 (mínima) al 10 (máxima), siendo el valor promedio 8,4. La aparición de colapso circulatorio o pérdida conciencia en esta serie era excepcional18. La necrosis de la pared intestinal no ha sido descrita, si bien el edema de la pared intestinal puede originar crisis de oclusión intestinal. Si se realizan estudios de imagen durante los ataques se pueden observan imágenes en «huella dactilar» o espiculación del asa afecta19.
En el caso de nuestro paciente, dada la intensidad del dolor y la afectación del estado general, se procedió a la realización en primer lugar de una ecografía y posteriormente de un TAC abdominal sin contraste intravenoso, por la leve disfunción renal, para descartar otros procesos intraabdominales, especialmente la perforación de alguna víscera hueca. La ausencia de fiebre durante las crisis de dolor en pacientes afectos de AH y la adecuada selección de técnicas de imagen, orientan al clínico para evitar laparotomías en blanco.
Las causas mas frecuentes en nuestro medio de PA son el consumo de alcohol y la litiasis biliar. Nuestro paciente no consumía alcohol y los estudios de imagen mediante resonancia nuclear magnética y ecoendoscopia descartaron la presencia de microlitiasis o barro biliar, así como de datos que orientaran a anomalías en los conductos de drenaje pancreáticos o pancreatitis crónica.
Se estima que el 3–5% de las PA son de origen farmacológico, siendo los agentes más frecuentemente implicados: azatioprina, mesalazina y didanosina. La mayoría de los casos publicados se tratan de series cortas o casos aislados. Nuestro paciente llevaba tomando 10 mg de enalapril desde hacía seis años; este antihipertensivo es definido como medicamento de clase ii atendiendo a Trivedi y Pitchumoni20, que tras una revisión pormenorizada en la literatura de los fármacos asociados a PA desde 1966 hasta 2004, incluyen en esta clase a aquellos medicamentos de los que solo se habían informado menos de veinte casos (en concreto con el enalapril doce casos). Nuestro paciente había presentado en las horas previas al episodio de pancreatitis lesiones típicas de AH en miembros superiores y la reexposición durante tres meses no había originado un nuevo ataque de pancreatitis, por lo que es poco probable en este contexto una relación causa-efecto, siendo más plausible el origen en su enfermedad de base. El enalapril fue retirado posteriormente a los tres meses del alta hospitalaria dada la posibilidad de originar crisis de angioedema, a través de un mecanismo de interacción con la calicreína21.
Los niveles en sangre de calcio, y triglicéridos estaban en el rango de la normalidad por lo que se descartaron las causas metabólicas. Los anticuerpos antinucleares eran negativos y los niveles de IgG4 era normales por lo que se descartó pancreatitis autoinmunitaria22.
El mecanismo fisiopatológico de la PA en la AH es probable que esté en relación con la aparición de edema a nivel pancreático y/o periampular que dificulte el drenaje del conducto de Wirsung5,6. Otro mecanismo posible sería una mutación genética en el gen regulador transmembrana de la fibrosis cística y en los genes del tripsinogeno catiónico23.
El establecimiento de la etiología de la PA no solo tiene implicaciones en el tratamiento del proceso inflamatorio, sino también en la prevención de las recurrencias24,25. El momento y la indicación de la realización de pruebas invasivas como la ecoendoscopia en el caso de la PA idiopática no está totalmente aclarada, si bien su rentabilidad diagnóstica oscila entre el 60 y 80%. En la serie de Repiso et al26, no se apreciaban diferencias en la rentabilidad diagnóstica en cuanto a realizar la ecoendoscopia tras el primer episodio (48%) o tras episodios recurrentes (37%), por lo que es probable que al no retrasar la exploración se disminuyan las recurrencias. En nuestro caso se optó por realizarla tras el primer episodio al haber precisado el paciente ingreso en la unidad de cuidados intensivos debido a la disfunción renal.
El empleo de antihistamínicos, epinefrina o esteroides no han demostrado ser efectivos durante la crisis de angioedema, siendo el tratamiento de elección el empleo de C1 inhibidor o en su defecto plasma fresco congelado, si bien la administración de este último es controvertido, por la posibilidad de empeorar el angioedema al contener dicho plasma C2 y C427. Respecto al tratamiento profiláctico en los periodos intercrisis, estan indicados andrógenos como el danazol o el estanozolol al estimular la síntesis hepática de C1 inhibidor, si bien presentan importantes efectos secundarios, entre ellos: virilización, formación de adenomas hepatocelulares, hepatitis colestásica, PA, retraso de crecimiento… por lo que se emplean fundamentalmente en la profilaxis a corto plazo de procedimientos médicos tales como extracciones dentales, endoscopias, intubaciones orotraqueales , que pueden precipitar episodios de angioedema1,28,29. Por otra parte los agentes antifibrinolíticos (ácido tranexámico, ácido epsilón aminocaproico) son de primera elección en los pacientes pediátricos y en mujeres embarazadas, dados los efectos deletéreos de los andrógenos sobre el feto: virilización, alteración en su desarrollo y cierre prematuro de la placa epifisiaria30,31. Se ha observado que tras 28 días de tratamiento con cualquiera de los dos agentes, andrógenos o antifibrinolíticos, se reduce un 90% la frecuencia de ataques de angioedema. No hay estudios comparativos entre ambos fármacos, pero parece que el uso de anabolizantes es superior1.
En el caso de nuestro paciente, le fue prescrito ácido tranexámico a dosis de 20mgr/kg/d, dado que ya había rechazado previamente el empleo del danazol por sus efectos secundarios, además de haberse sido descrito episodios de PA secundarios al empleo de este tratamiento32. Tras doce meses de seguimiento permanece asintomático sin nuevas crisis de dolor abdominal ni de angioedema.
En resumen, presentamos el caso de un paciente afecto de AH tipo 2, que desarrolla como complicación excepcional una PA. Dada la frecuencia de episodios de dolor abdominal recurrente en pacientes con AH, es recomendable además de una adecuada historia clínica, la determinación de amilasa o lipasa, por las implicaciones pronósticas y terapéuticas de la PA. En nuestro conocimiento se trata del séptimo caso descrito en la literatura mundial de PA asociada a AH.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.

