




Las porfirias agudas (PA) son trastornos hereditarios en la síntesis del grupo hemo que se caracterizan por la aparición de episodios agudos de dolor abdominal, crisis de hipertensión arterial, taquicardia y trastornos neuropsiquiátricos, llegando incluso a provocar convulsiones, parálisis ascendente o coma. El retraso o error en el diagnóstico puede empeorar gravemente el pronóstico. Presentamos el caso de una paciente con porfiria aguda intermitente (PAI) subclínica y hepatitis crónica diagnosticada de forma casual por una elevación de transaminasas en el estudio analítico.
Acute porphyria is a term that encompasses a group of hereditary disorders involving defects in heme metabolism, characterized by acute episodes of abdominal pain, acute hypertension, tachycardia and neuropsychiatric disorders, sometimes leading to convulsions, ascending paralysis and coma. Misdiagnosis or delayed diagnosis can seriously worsen prognosis. We report the case of a woman with subclinical acute intermittent porphyria and chronic hepatitis incidentally diagnosed due to transaminase elevation on laboratory analysis.
Las porfirias agudas (PA) son trastornos hereditarios que se caracterizan por la aparición de episodios agudos de dolor abdominal, crisis de hipertensión arterial, taquicardia y trastornos neuropsiquiátricos, llegando incluso a provocar convulsiones, parálisis ascendente o coma. El retraso o error en el diagnóstico puede empeorar gravemente el pronóstico. Presentamos el caso de una paciente con porfiria aguda intermitente (PAI) subclínica y hepatitis crónica. Comentamos la dificultad diagnóstica, el pronóstico de estos pacientes y el seguimiento recomendado en la literatura médica en cuanto a la enfermedad hepática.
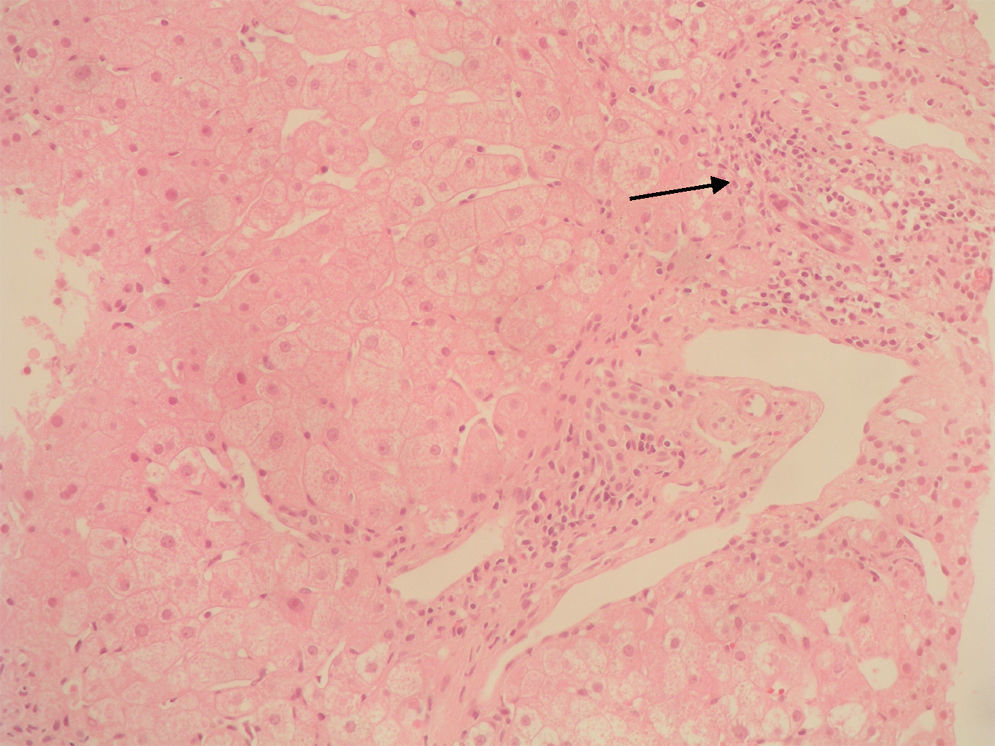
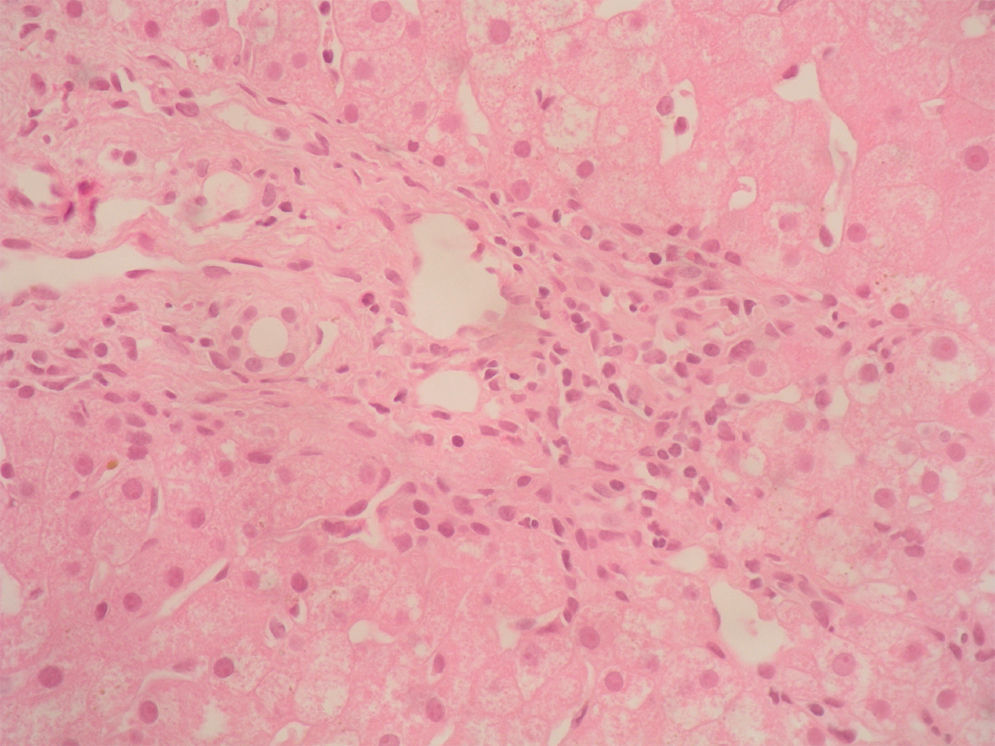
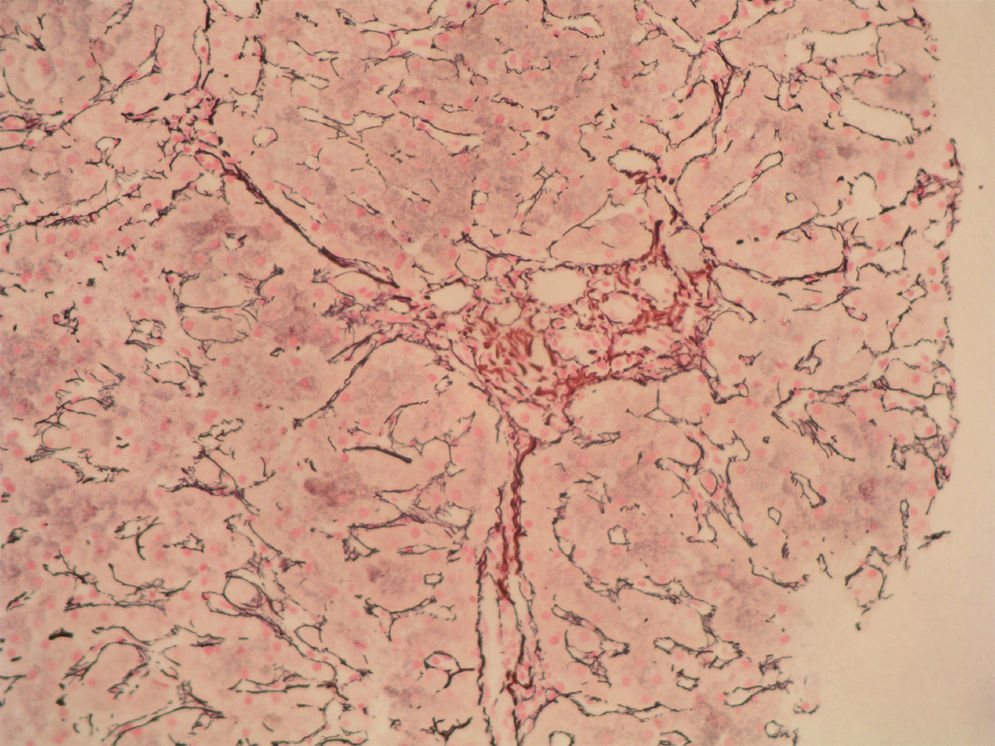
Caso clínicoSe trata de una mujer de 33 años de edad que se remite a consulta para estudio por elevación crónica de transaminasas, detectada de forma casual. Entre sus antecedentes familiares destaca una prima hermana diagnosticada de crisis de PAI y madre, tío materno y varios primos diagnosticados de PAI subclínica. La paciente niega antecedentes personales de interés o hábitos tóxicos, así como la existencia de sintomatología alguna. En la exploración física no se encuentran alteraciones. El análisis hematológico y bioquímico fue normal, salvo los valores de GOT/GPT que oscilaban entre 48/44 y 52/58 U. La bilirrubina, fosfatasa alcalina (FA) y GGT también fueron normales. Se determinaron también los valores de TSH y de hierro en sangre que fueron normales. La serología para virus de hepatitis B y C resultó negativa. Los anticuerpos antinucleares (ANA), antimitocondriales (AMA) y anti-LKM fueron negativos, detectándose valores positivos de anticuerpos antimúsculo liso (AML), con cifras de 1/160. Se obtuvo un valor elevado de IgG, en torno a 1.640 U. Los anticuerpos antitransglutaminasa, alfa-1-antitripsina y la ceruloplasmina, así como la excreción de cobre en orina de 24h eran también normales. Un primer análisis de orina mostró valores de porfirinas dentro de límites normales. Se realizó además una ecografía abdominal sin encontrar alteraciones. Ante la imposibilidad de llegar a un diagnóstico y habiéndose descartado otras causas probables de hipertransaminasemia, se hizo una biopsia hepática en la que se obtuvo un cilindro con arquitectura trabecular hepática conservada, un infiltrado linfocitario que sobrepasaba la interfase y algunos tractos fibrosos porto-portales (figs. 1–3). Estos hallazgos corresponden una fibrosis en estadio 2 de la escala Metavir, compatible con hepatitis autoinmune (HAI). En función de estos resultados y según los criterios diagnósticos expuestos por Hennes et al, donde se obtuvo una puntuación de 6, el diagnóstico de hepatitis autoinmune es probable17. Por ello, se decidió instaurar tratamiento inmunosupresor combinado con prednisona 40mg y azatioprina hasta 100mg al día. Durante el seguimiento no se evidenció respuesta bioquímica, incluso se produjo un leve aumento de los valores de GOT/GPT hasta cifras de 74/92. En este punto de la evolución parece muy improbable el diagnóstico de HAI, por lo cual se retiró el tratamiento. Posteriores determinaciones de anticuerpos anti-AML fueron negativas. Durante este tiempo de seguimiento la madre se realiza estudio específico de actividad de la enzima PGB deaminasa (porfobilinogenodeaminasa) en el mismo centro que sus familiares, diagnosticándose también PAI subclínica. Teniendo en cuenta este antecedente familiar directo, propusimos a la paciente un estudio de porfirinas más completo en otro centro, detectándose en esta ocasión una excreción de uroporfibilinógeno y ácido delta-aminolevulínico en orina discretamente por encima de los valores normales, porfirinas en heces normales y actividad de la enzima PGB desaminasa en eritrocitos francamente disminuida, con valores de 42 U/LH (N: 86-160). Finalmente establecimos el diagnóstico de PAI subclínica con hepatitis crónica secundaria a la misma. En la actualidad la paciente permanece asintomática con valores de GPT casi normales y en seguimiento en consultas externas de digestivo.
 de una muestra hepática donde aparece un infiltrado inflamatorio linfocitario, en los espacios porta (flecha).")
.")

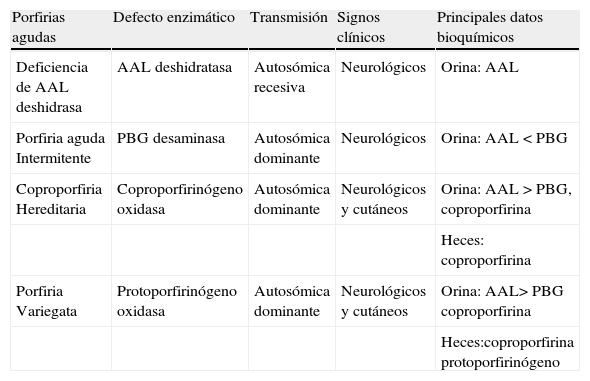
Las PA son errores hereditarios causados por deficiencia enzimática parcial en la biosíntesis del grupo hemo (tabla 1). Son de herencia autosómica dominante y como ya se ha dicho anteriormente, se manifiestan como episodios agudos de dolor abdominal, hipertensión arterial, taquicardia y trastornos neuropsiquiátricos, siendo estos cinco veces más frecuentes en el sexo femenino2. No obstante, la enfermedad cursa también con grandes periodos asintomáticos.
Clasificación de las porfirias agudas
| Porfirias agudas | Defecto enzimático | Transmisión | Signos clínicos | Principales datos bioquímicos |
| Deficiencia de AAL deshidrasa | AAL deshidratasa | Autosómica recesiva | Neurológicos | Orina: AAL |
| Porfiria aguda Intermitente | PBG desaminasa | Autosómica dominante | Neurológicos | Orina: AAL < PBG |
| Coproporfiria Hereditaria | Coproporfirinógeno oxidasa | Autosómica dominante | Neurológicos y cutáneos | Orina: AAL > PBG, coproporfirina |
| Heces: coproporfirina | ||||
| Porfiria Variegata | Protoporfirinógeno oxidasa | Autosómica dominante | Neurológicos y cutáneos | Orina: AAL> PBG coproporfirina |
| Heces:coproporfirina protoporfirinógeno |
AAL: ácido 5-aminolevulínico; PBG: porfobilinógeno.
La PAI se caracteriza por el déficit parcial de la enzima PBG desaminasa, la tercera enzima en la biosíntesis del hemo. Es el tipo de PA más frecuente y grave. Se transmite de forma autosómica dominante con un alto grado de heterogeneidad genética y más del 90% de los portadores permanecen asintomáticos a lo largo de su vida1. Afecta de 1-8/100.000 habitantes si consideramos una acumulación anormal de porfirinas, pero probablemente el trastorno está infraestimado1.En la actualidad, se ha incrementado el diagnóstico de casos asintomáticos basados en la medición de la actividad enzimática de la porfobilinógeno deaminasa4. Un estudio de análisis molecular sobre el déficit enzimático en donantes franceses caucasianos encontró una prevalencia del 0,6/1.000 habitantes, mucho más alta que la encontrada por la excreción aumentada de porfirinas en orina6. Las crisis agudas aparecen generalmente cuando factores precipitantes como el ayuno, el consumo de alcohol o tabaco, las infecciones o los fármacos aumentan su demanda o estimulan su síntesis por la vía del citocromo p450. Esto desencadena una producción excesiva de precursores como la alanina (ALA) y PBG, responsables a su vez de la toxicidad neurovisceral y del cuadro clínico. La coproporfiria hereditaria y la porfiria variegata además presentan lesiones dérmicas por aumento de precursores fotosensibles. El diagnóstico se realiza por un alto índice de sospecha clínica y la demostración de una excreción urinaria elevada de los metabolitos precursores, común durante los ataques3. Sin embargo, estos valores pueden ser normales durante los periodos asintomáticos y en pacientes prepúberes, lo cual dificulta el diagnóstico. Actualmente se ha incrementado el diagnóstico de casos asintomáticos basados en la medición de la actividad enzimática de la PBG desaminasa5. En contraste con la porfiria cutánea tarda y la protoporfiria, las PA no son asociadas habitualmente con enfermedad hepática crónica y por tanto no se contempla de manera habitual la PA asintomática como causa de elevación crónica de GPT7. No obstante, algunas series describen alteraciones de la GPT hasta en el 13% de los casos de PAI asintomáticos8.
La afectación hepática en las PA es variable y mal conocida. Podemos encontrar elevación de transaminasas sobre todo durante los ataques. Las biopsias hepáticas pueden mostrar esteatosis o siderosis pero lo más importante es un riesgo incrementado de desarrollar carcinoma hepatocelular (HCC). Desde la primera asociación descrita de PAI y HCC en 19849, varios estudios prospectivos de casos y controles sobre la mortalidad en este tipo de pacientes12 han puesto de manifiesto dicha asociación11–13, incluyendo casos de portadores asintomáticos. Comparando la incidencia de HCC en Francia, Andant et al estimaron en un estudio prospectivo 36 veces más riesgo de HCC que en la población general. Sin embargo, el 10% de los pacientes con PAI mueren a causa de la aparición de hepatomas13. La edad de aparición de HCC comienza a los 50 años siendo a mayor edad cuando se trata de casos asintomáticos probablemente debido a una menor acumulación de ALA en estos últimos. También la cirrosis hepática (HC) es más común entre los pacientes con PA (12 versus 0,5% en controles)10. Sin embargo, la existencia de cirrosis o fibrosis previa a la aparición de HCC no está clara y los resultados de las series son contradictorios. En un estudio retrospectivo en pacientes suecos los casos de HCC asientan en hígados ya cirróticos. No así en el estudio prospectivo realizado en la población francesa donde el hígado no tumoral fue normal en 5 de los 7 casos y cirrótico en los otros 2 restantes donde coexistía infección por VHB y VHC12.
En cuanto a la patogenia, parece que la acumulación de precursores como la ALA produce toxicidad directa oxidativa sobre el ADN por un aumento de radicales libres, de esta forma, los casos de HCC tienen significativamente más excreción de ALA que los controles sin HCC12. No conocemos los casos publicados en nuestro país de PAI subclínica con lesión de hepatitis crónica demostrada como ocurre en nuestro caso. Las muestras de biopsia hepática pueden mostrar esteatosis o depósito férrico y es fácil deducir que la mayoría de pacientes afectados de cirrosis han pasado antes por estadios de HC y distintos grados de fibrosis, pero hay pocos datos publicados que incluyan biopsias hepáticas y menos aún en casos subclínicos. Habría que considerar las PA subclínicas en casos de enfermedad hepática de etiología incierta, incluyendo HC y cirrosis criptogénicas. La confusión con HAI (como ocurrió inicialmente en nuestro caso) es fácil, teniendo en cuenta que las dos enfermedades son más frecuentes en mujeres jóvenes y los autoanticuerpos pueden aparecer de forma inespecífica a títulos bajos de forma transitoria o bien tratarse de pacientes con HAI y autoanticuerpos convencionales negativos14–16. La biopsia hepática rara vez es específica y como en nuestro caso puede ser compatible con el diagnóstico de HAI. En la mayoría de los pacientes no se incluyen las PA en el diagnostico etiológico de hepatitis crónica o cirrosis. Incluso las recomendaciones de programas de vigilancia de pacientes con riesgo de HCC no incluyen a los pacientes con PA17. La exclusión de otras causas de enfermedad hepática y unos antecedentes familiares compatibles nos deben hacer pensar en esta enfermedad hereditaria y determinar el déficit de actividad enzimática en un laboratorio cualificado si los resultados en orina no fuesen patológicos como es lo más habitual. Por tanto, consideramos importante tener en cuenta el diagnóstico de PA en pacientes con elevación de transaminasas y/o hepatopatías de origen no filiado. De la misma forma, debemos considerar la existencia de portadores asintomáticos en estudios de familiares de PA. Harían falta más estudios para determinar qué grupo de pacientes con PA tienen realmente un riesgo relativo de HCC aumentado de forma significativa.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.

