




La metilación del ADN se ha revelado como un mecanismo epigenético fundamental en la regulación de la expresión de genes que controlan funciones celulares cruciales en el desarrollo del cáncer. Defectos en la metilación (tanto hipometilación global como hipermetilación de islas CpG) están implicados en la carcinogénesis colorrectal. Algunos nutrientes ejercen un claro efecto en la metilación, lo que sugiere que algunas diferencias en la incidencia de cáncer colorrectal asociadas a la dieta podrían ser debidas al efecto de esta en la metilación. La presencia de los defectos en la metilación tiene claras implicaciones diagnósticas y de pronóstico. Así, se están ya ensayando diversos tests para el cribado del cáncer colorrectal basados en el análisis de genes metilados bien en heces bien en sangre. Por otro lado, la reversibilidad de los procesos de metilación permite el desarrollo de quimioterapias que regulan este proceso a través de su actividad antineoplásica.
DNA methylation is a fundamental epigenetic mechanism in regulating the expression of genes controlling crucial cell functions in cancer development. Methylation defects (both global hypomethylation and hypermethylation of CpG islands) are implicated in colorectal carcinogenesis. Some nutrients have a clear effect on methylation, suggesting that some dietary-associated differences in the incidence of colorectal cancer could be due to the effect of diet on methylation. The presence of methylation defects has clear diagnostic and prognostic implications. Thus, several tests are being used for colorectal cancer screening based on methylated gene analysis, whether in feces or blood. In addition, the reversibility of methylation processes allows the development of chemotherapies that regulate this process through their antineoplastic activity.
En los últimos años se ha puesto de relevancia la importancia de la epigenética. La epigenética se basa en mecanismos celulares que introducen variaciones en la expresión génica sin afectar la secuencia del ADN. Entre estos mecanismos destacan la modificación de la estructura de la cromatina a través de la modificación de histonas, los mecanismos de acción de los miARN, y la metilación. Esta última es un proceso reversible en el cual se introduce o elimina un grupo metilo (-CH3) en las citosinas presentes en dinucleótidos CpG en el genoma. Estos dinucleótidos se concentran en zonas llamadas islas CpG y frecuentemente se localizan en los promotores génicos (regiones del ADN que facilitan la transcripción de los genes). Aproximadamente el 60% de los genes humanos contienen islas CpG en la zona 5’ de su promotor1.
La metilación del ADN es un mecanismo habitual de control de diferentes mecanismos celulares como la protección de la integridad del genoma, el mantenimiento de patrones de expresión específicos de cada tejido o la definición de la impronta genética. La distribución de estos patrones de metilación es específica de cada tipo celular y se establece durante el desarrollo embrionario. La alteración de los patrones de metilación del ADN se han descrito en varias enfermedades y en muchos tipos de cáncer. Esta alteración puede ser por un incremento o hipermetilación que conlleva una represión de la transcripción génica, o por una disminución o hipometilación, con una consecuente activación de esta transcripción. Se ha demostrado que la hipometilación del ADN precede a la inestabilidad genética en los cánceres gastrointestinales. El grado de hipometilación del genoma parece correlacionarse con el nivel de alteraciones cromosómicas y puede afectar la estabilidad de los cromosomas2, los mecanismos de impronta genética y la activación de genes normalmente silenciados3. El ADN tumoral en general está hipometilado, sin embargo la hipermetilación afecta genes específicos en un contexto general de hipometilación. La hipermetilación de promotores génicos de genes supresores inactiva su transcripción, facilitando así el desarrollo del tumor.
En los últimos años el interés sobre el estudio de la metilación del ADN en el cáncer colorrectal (CCR) ha ido incrementándose. Así, se han descrito alrededor de 50 genes con alteraciones somáticas del grado de metilación en distintos estadios del CCR identificándose así como un potencial biomarcador. Al ser la metilación un proceso reversible, la industria farmacéutica está invirtiendo importantes recursos en el desarrollo de fármacos como agentes demetilantes de genes oncosupresores. Al ser la metilación un fenómeno que se ve influenciado por la dieta, también se hace evidente el potencial uso de nutrientes como agentes quimiopreventivos.
En este artículo repasaremos estos aspectos en el CCR esporádico y también detallaremos las alteraciones en la metilación descritas más recientemente en casos de CCR hereditario.
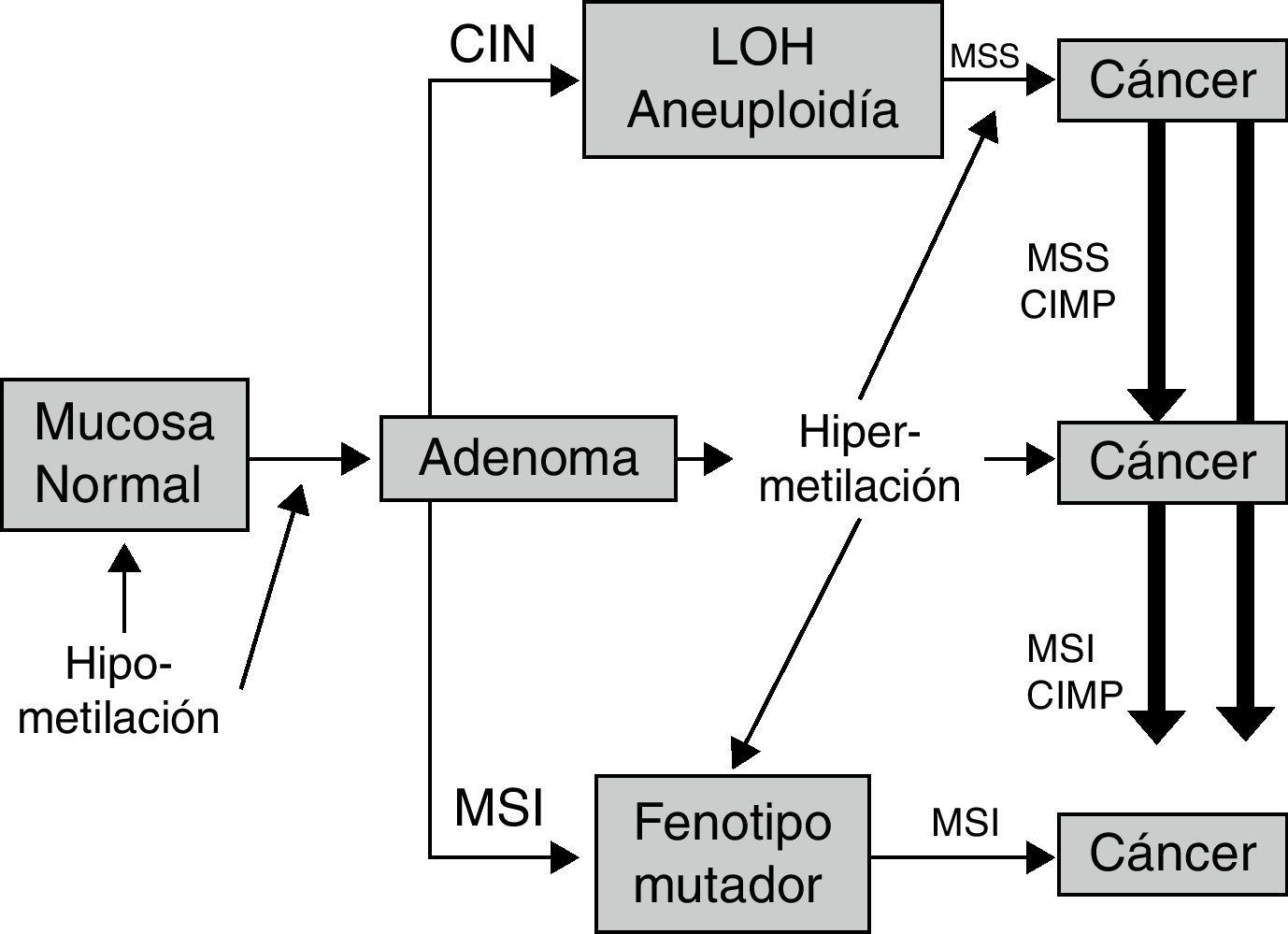
Cáncer colorrectal esporádicoMuchos de los CCR se desarrollan a través de un proceso con múltiples etapas, conocido como la secuencia adenoma-carcinoma, propuesto por Fearon y Vogelstein hace más de 2 décadas. Durante este proceso se van acumulando alteraciones genéticas y epigenéticas en genes cruciales en el control del crecimiento y la diferenciación celular. La inestabilidad genómica es característica del CCR y afecta a todo el genoma, implicando la pérdida de la integridad del ADN lo cual acelera el proceso de acumulación de mutaciones. Esta inestabilidad puede ser cromosómica (CIN) o de microsatélites (MSI). La presencia tanto de CIN como de MSI se ha descrito en estadios iniciales del desarrollo de CCR identificándose incluso en adenomas4,5. Los tumores con CIN son los más comunes y presentan pérdida de heterocigocidad, frecuentes alteraciones citogenéticas y pérdida alélica6. Este proceso es conocido como la vía supresora del CCR. Los tumores que presentan CIN también muestran inactivación de genes oncosupresores como APC, p53, DCC, SMAD2 y SMAD4, sin embargo, los genes que permiten el establecimiento de esta inestabilidad aún no están bien establecidos. Los tumores con MSI se desarrollan como resultado de la alteración del sistema del Mismatch Repair (MMR) o de reparación de mal apareamientos. La función de las proteínas codificadas por los genes de este sistema (MLH1, MSH2, MSH3, MSH6, PMS2) es reparar los errores de apareamiento entre bases que se producen habitualmente y de forma periódica durante la replicación del ADN7. Cuando estos genes no funcionan, se acumulan mutaciones a lo largo del genoma especialmente en secuencias repetitivas de nucleótidos conocidas como microsatélites. Estos últimos se localizan en zonas no codificantes y también en secuencias codificantes de genes que controlan importantes mecanismos celulares como IGFR2, TGFβR2, BAX o MSH6. La MSI se identifica en un 10-15% de los CCR. En la gran mayoría de estos casos la presencia de MSI se debe a la hipermetilación del promotor del gen MLH1 con la consecuente inactivación de la transcripción génica y la pérdida de la expresión proteica8. La alteración de la metilación de MLH1 es una de las evidencias más sólidas de la implicación de defectos de metilación en el desarrollo del CCR y es la responsable de un 80% de casos con MSI. Sin embargo, otros genes como MGMT, p16, p14 también se encuentran metilados en subgrupos tanto de CCR como de adenomas9,10. La figura 1 representa las distintas vías carcinogénicas descritas.
![Vías carcinogénicas del cáncer colorrectal. Las 2 principales vías carcinogénicas en el cáncer colorrectal son la vía supresora (caracterizada por la pérdida de heterocigicidad [LOH] y aneuploidia), y la vía mutadora (caracterizada por la inestabilidad de microsatélites o MSI). Defectos de la metilación han sido implicados tanto en estadios iniciales como más avanzados.](https://static.elsevier.es/multimedia/02105705/0000003500000007/v1_201305141419/S0210570512000544/v1_201305141419/es/main.assets/gr1.jpeg?xkr=ue/ImdikoIMrsJoerZ+w997EogCnBdOOD93cPFbanNcRcp7AE8pxI7/lU7i5mKGl6o67DY/uL2+/IxZemV5MIshc00TFNCinfT21eHzPhjgbVZNW7aiV2C4SOtpRRBZoWlykqYjzaHi7jQNPCfb8vccjK5Yo+1f5qo3/mNJkMG271ObNndPozx3/sFBjCAmCgSMXv/yMuOfsmyZH8kk390mHnhbixBRdIOFJvyBSbCdM9jXuNdVpFDyh6d83PgxXCp3mxK0vy3QFkcBT0In36/cTGH+15hZgYnrVvvhFHmU= "Vías carcinogénicas del cáncer colorrectal. Las 2 principales vías carcinogénicas en el cáncer colorrectal son la vía supresora (caracterizada por la pérdida de heterocigicidad [LOH] y aneuploidia), y la vía mutadora (caracterizada por la inestabilidad de microsatélites o MSI). Defectos de la metilación han sido implicados tanto en estadios iniciales como más avanzados.")
Vías carcinogénicas del cáncer colorrectal. Las 2 principales vías carcinogénicas en el cáncer colorrectal son la vía supresora (caracterizada por la pérdida de heterocigicidad [LOH] y aneuploidia), y la vía mutadora (caracterizada por la inestabilidad de microsatélites o MSI). Defectos de la metilación han sido implicados tanto en estadios iniciales como más avanzados.
Recientemente se ha descubierto que esta hipermetilación también puede afectar la transcripción génica de manera indirecta, regulando la expresión de los miARN. Los miARN son pequeñas moléculas de ARN no codificante que regulan la traducción de transcritos génicos por unión complementaria a estos o induciendo su eliminación. Estos pueden funcionar como supresores o promotores de la carcinogénesis y debido a su pequeño tamaño tienen el potencial de controlar muchos genes distintos gracias a una alta complementariedad. Diversos trabajos han descrito la implicación de este tipo de regulación en diferentes cánceres, incluyendo el CCR, así como en los procesos metastásicos11. La alteración de los patrones de metilación en el CCR también afecta la regulación de la transcripción de estas moléculas. Este mecanismo parece ser el causante, por ejemplo, de la represión de la transcripción de la familia de miR-34 que regula la expresión de p53, un gen oncosupresor frecuentemente implicado en distintos cánceres, incluido el CCR12.
Fenotipo metilador de islas CpG en el cáncer colorrectalToyota et al. describieron por primera vez un grupo de tumores con un fenotipo metilador de islas CpG o CIMP. Estos tumores presentaban un alto grado de metilación de marcadores específicos y tenían MSI debido a que MLH1 también estaba metilado13. A este fenotipo se le han asociado distintas características como una localización proximal del tumor, producción de mucina o ser más prevalente en tumores de mujeres de avanzada edad. Sin embargo, estas características están en realidad asociadas a tumores MSI. Al igual que en los casos MSI, los pacientes con CCR con alto grado de metilación en islas CpG tampoco parecen beneficiarse de quimioterapia con 5-fluorouracilo14.
La entidad de CIMP como un tipo diferente de inestabilidad genómica al mismo nivel que CIN o MSI es aún un tema de controversia. Los tumores CIMP se asocian habitualmente a mutaciones en BRAF y presencia de MSI, pero también existen tumores CIMP con estabilidad de microsatélites (MSS) y mutaciones en KRAS en lugar de BRAF15. Es posible que la frecuente identificación de CIMP con MSI sea debida a los marcadores comúnmente analizados o incluso a la tecnología empleada hasta la fecha. De hecho, este fenotipo se describió con un conjunto de marcadores que estaban altamente hipermetilados (CDKN2A, MINT1, MINT2, MINT31 y MLH1). Más tarde se han propuesto otros paneles más específicos y sensibles para identificar CIMP sin inestabilidad de microsatélites16. Hasta 4 paneles distintos de marcadores han sido descritos, lo que ha contribuido a introducir variabilidad en la identificación de estos tumores. Así pues aún está por determinar si el fenotipo CIMP es una entidad por sí misma o si la metilación es un efecto secundario del proceso carcinogénico. Quizá los análisis de la totalidad del metiloma acaben determinando este hecho con mayor certeza.
Avances en el conocimiento de la metilación génica en el cáncer colorrectalLos primeros análisis de metilación se basaban en la cuantificación de la cantidad total de citosinas metiladas en el genoma. Sin embargo, al identificarse la hipermetilación de promotores génicos se creó la necesidad de analizar específicamente estas regiones. Distintas estrategias has sido utilizadas para detectar la metilación del ADN lo que ha resultado en un amplio abanico de metodologías17. Las 3 principales estrategias se basan en: a) digestión del ADN con enzimas de restricción sensibles a cambios en los patrones de metilación; b) modificación química de las citosinas metiladas utilizando bisulfito; y c) la purificación de la fracción metilada del genoma a través de anticuerpos. Las técnicas moleculares para evaluar los patrones de metilación han evolucionado progresivamente hacia la identificación y cuantificación de regiones específicas en el genoma. Sin embargo, como en el caso del análisis de la secuencia del ADN a nivel genómico, ahora también existe la posibilidad de analizar el metiloma a través de arrays o de secuenciación de nueva generación a gran escala. El desarrollo de estas aplicaciones con más resolución ha permitido el descubrimiento de nuevos genes alterados y la comprobación de la implicación de genes ya identificados. Así, Oster et al. analizaron el metiloma de tumores MSI, tumores MSS y adenomas. Utilizando arrays de metilación se analizaron 14.000 genes, de los cuales 95 (0,68%) mostraban una metilación de las islas CpG significativamente diferente en comparación con las muestras normales en adenomas y carcinomas. Sesenta y cuatro de estos estaban hipermetilados y el resto hipometilados. Quince de los genes hipermetilados ya habían sido descritos y algunos eran genes incluidos en los paneles de marcadores de CIMP, diferenciando los tumores MSI. Un total de 92 genes diferenciaban significativamente los tumores MSS y MSI. Paralelamente, este análisis permitió la identificación de nuevos genes potencialmente relacionados con el desarrollo del CCR, como JAM2 o NRG1, los cuales mostraban una fuerte correlación inversa entre la metilación y la expresión génica18.
Las nuevas tecnologías también permiten la introducción de un nuevo conocimiento de mecanismos alternativos a los conocidos, como es el caso de la definición de los «shores» de las islas CpG en CCR. Irizarry et al. recientemente propusieron que las zonas genómicas más relevantes para el control de la transcripción génica se localizan a 2kb de las islas CpG. Los cambios de los patrones de metilación asociados con el proceso carcinogénico se producen en estos «shores», que en una situación celular normal muestran los patrones de metilación relacionados con la diferenciación específica del tejido. Esta teoría avalaría la idea de que el desarrollo del cáncer es en parte el resultado de una perturbación en los mecanismos que mantienen la diferenciación de los tejidos19.
El nuevo conocimiento que está surgiendo como resultado del avance de las nuevas tecnologías será crucial para el diseño e implementación de estudios de asociación del epigenoma (EWAS) a gran escala, como ocurrió con los GWAS, que empezarán a publicarse en los próximos años20.
Biomarcadores de metilación en el cribado del cáncer colorrectalEl CCR es uno de los cánceres comunes que más puede prevenirse dado su lento desarrollo y su excelente pronóstico cuando es detectado en estadios iniciales. Una de las técnicas más establecidas para el cribado poblacional es la preselección de individuos para la realización de colonoscopias mediante el uso de tests de sangre oculta en heces. Esta técnica no es invasiva, pero muestra un valor predictivo bajo y su capacidad de reducción de mortalidad es controvertida21. Basándose en estos datos, países como los Estados Unidos han apostado ampliamente por la colonoscopia como método de cribado poblacional. Esta aproximación, aparte de ser más invasiva y contar con un pequeño riesgo de complicaciones, también tiene sus limitaciones, ya que requiere un gran número de endoscopistas y muestra una escasa eficacia en la identificación de tumores en el colon derecho22. Por todos estos motivos, continúa siendo muy necesaria la identificación de técnicas sencillas, no invasivas, y que tengan una alta sensibilidad y especificidad para el diagnóstico de cánceres iniciales y lesiones premalignas. En este sentido se han empezado a estudiar biomarcadores de metilación en heces o suero/plasma. En el primer método se analiza la metilación del ADN proveniente de la exfoliación de células colónicas, y en el segundo, se identifica la metilación del ADN tumoral circulante que ha pasado a la sangre procedente del tumor.
Entre 2004 y 2009 se publicaron 19 trabajos en los que se describía la utilidad del análisis de metilación de diferentes paneles de genes en CCR y adenomas en ADN de heces. En general, la sensibilidad y especificidad para detectar una neoplasia colorrectal eran aproximadamente del 62 y el 89%23, respectivamente. Estos datos son mucho más esperanzadores que el test de guayaco o de sangre oculta en heces (FOBT) que muestra una sensibilidad menor del 15% para la detección de una neoplasia colorrectal con una especificidad del 90%24. Sin embargo, aún no está clara la eficacia de estos tests de metilación del ADN en comparación con los tests inmunoquímicos fecales de nueva generación, los cuales muestran una clara superioridad respecto a los FOBT25.
Entre los genes estudiados como marcadores metilados se incluyen casos ya bien establecidos como MLH1 o MGMT, así como nuevos candidatos como el gen Vimentin, que se encuentra frecuentemente metilado en ADN de heces de pacientes con CCR pero raramente en heces procedente de individuos sanos.
En el campo de la utilización de tests para metilación de ADN en suero o plasma, si bien estos parecen que tienden a mostrar una sensibilidad y especificad parecida a los tests en heces, los trabajos publicados aún son escasos. En cualquier caso, algunos estudios también sugieren un potencial uso como marcadores de pronóstico y seguimiento de la enfermedad26,27.
En resumen, el uso de marcadores de metilación para el cribado del CCR podría ser una realidad, aunque para eso aún carecemos de estudios a gran escala que avalen esta aproximación. Estos estudios deberán incluir a pacientes con tumores de diferentes estadios, pacientes con lesiones premalignas, individuos sanos y pacientes con otros cánceres comunes para demostrar su utilidad como marcadores eficaces.
Cáncer colorrectal hereditarioEl síndrome de Lynch es la forma hereditaria más común de CCR. Este es un síndrome autosómico dominante causante de múltiples cánceres y que afecta al 1-3% de los pacientes con CCR con una penetrancia de hasta el 80% para este cáncer28. Estos pacientes no muestran características clínicas que adviertan sobre el alto potencial de cáncer, a diferencia de las formas hereditarias de CCR con poliposis29. El síndrome de Lynch está causado por mutaciones patogénicas en los genes del sistema del MMR: MLH1, MSH2, MSH6 y PMS2. El 90% de los casos muestran mutaciones en MLH1 o MSH2. Como resultado de estas mutaciones los tumores de estos pacientes muestran MSI y normalmente pérdida de expresión de la proteína del gen afectado. Dependiendo del gen alterado, estos pacientes muestran un riesgo de CCR y de cánceres extracolónicos distinto30. Estos pacientes necesitan un manejo médico adecuado, basado en estos riesgos31. En la actualidad el diagnóstico de este síndrome se efectúa de forma molecular al identificar a un individuo con una mutación patogénica en alguno de estos genes. Como ya se describito en este artículo, la implicación de los defectos de metilación en el desarrollo del CCR esporádico está bien establecida, sin embargo, hasta la aparición de estudios recientes no se ha empezado a implicar estos procesos en casos sindrómicos. De hecho, diversos trabajos han asociado defectos de metilación en casos hereditarios con un fenotipo similar al del síndrome de Lynch. Estos defectos se conocen como epimutaciones constitutivas y se traducen en una hipermetilación de promotores génicos con la consecuente silenciación de la transcripción de uno de los alelos en los tejidos somáticos normales no transformados.
En el año 2002 se publicó el primer trabajo describiendo un caso de metilación del gen MLH1 en la línea germinal de una paciente con CCR de 25 años. Esta paciente presentaba un tumor con un fenotipo MSI con pérdida de expresión de MLH132. El análisis genético de los genes MMR no identificó ninguna mutación germinal, pero sí se identificó hipermetilación del promotor de MLH1 en linfocitos periféricos. Además, esta paciente presentaba el polimorfismo c.93G>A (rs1800734) en heterocigosis, permitiendo de esta manera la discriminación de cada alelo. Así se pudo determinar que la metilación solo afectaba al alelo con el polimorfismo, confirmando que la paciente presentaba una metilación monoalélica.
Más tarde, otras publicaciones han reportado casos similares en pacientes de distintos orígenes con fenotipos Lynch. Sin embargo, estos casos no presentan una historia familiar de cáncer tan enriquecida como en las familias que tienen síndrome de Lynch con una mutación germinal en la secuencia de un gen MMR. De hecho, las epimutaciones en varios de estos pacientes se han descrito como de novo y por ese motivo estos pacientes no presentan historia familiar de cáncer. Sin embargo, en 2 de estos pacientes se ha demostrado la transmisión de la alteración a la siguiente generación, pero sin mostrar un patrón de herencia mendeliano. En una de estas familias, uno de los 3 hijos de la paciente mostraba la misma epimutación en la línea germinal, pero no en sus gametos. Esto probablemente es debido a que las epimutaciones en el ADN muestran una naturaleza mucho más lábil que las mutaciones puntuales y pueden revertirse entre generaciones. La transmisión hereditaria de estos defectos en islas CpG y las modificaciones en la cromatina parecen improbables, debido a la reprogramación epigenética que se produce durante la reproducción en mamíferos33. Hasta la fecha se han descrito 24 pacientes con epimutaciones en MLH1 en la línea germinal. Solo 4 de ellos son altamente sospechosos de síndrome de Lynch. Sin embargo, la mitad de estos pacientes presenta cánceres extracolónicos asociados con este síndrome34.
Con estos datos, y a falta de más estudios, los pacientes con epimutaciones constitutivas en MLH1 deben seguir las mismas pautas de manejo y seguimiento que un paciente con una mutación patogénica en la secuencia del DNA. A los familiares de primer grado se les debe ofrecer análisis molecular para determinar si son portadores de la epimutación, aunque el riesgo de cáncer de estos familiares parece ser menor del 50%.
En el año 2006, Chan et al.35 describieron a una familia que presentaba una epimutación en el gen MSH2. Tres hermano/as de la familia habían desarrollado CCR MSI y de endometrio antes de los 50 años y presentaban pérdida de expresión de la correspondiente proteína. El probando presentaba metilación del promotor en la mucosa colónica normal y tumoral. No se identificó ninguna mutación en la secuencia del gen. En 12 familiares se analizó la metilación del promotor y se genotiparon SNP informativos de la zona génica de MSH2. En 10 de 12 familiares de distintas generaciones se identificó metilación y la presencia de un haplotipo específico, evidenciando la presencia de una alteración genética en cis. Otra característica de estos individuos era el nivel variable de metilación. En el tejido colónico la metilación era muy abundante, pero en linfocitos periféricos no. Además, el grado de metilación variaba también entre individuos. Este mosaicismo, y el hecho de una clara transmisión entre generaciones, diferenciaba este mecanismo del que hemos descrito para MLH1. Tres años más tarde, un grupo holandés identificó esta alteración genética en cis36. En la región 5’ de MSH2 se encuentra el gen EPCAM. En estas familias, este gen presenta una deleción de los últimos exones donde se encuentra la señal de poliadenilación del ARN mensajero. La pérdida de esta señal elimina la terminación de la transcripción y se traduce en una elongación del transcrito de EPCAM en MSH2, creando una fusión entre los 2 transcritos. Sin embargo, aún no se conoce el mecanismo por el cual se activa la metilación de MSH2 con su consecuente pérdida de expresión. La metilación es secundaria a la mutación genética en EPCAM, así que estas mutaciones se suman al conjunto de alteraciones causantes del síndrome de Lynch. Por lo tanto, el análisis de EPCAM ya forma parte de la estrategia de diagnóstico del síndrome de Lynch cuando hay pérdida de expresión de MSH2 sin mutación germinal en este gen. Estas mutaciones explican el 1-3% de los casos con este fenotipo hereditario37.
Otro tipo de defecto de metilación que afecta a la mucosa colónica tumoral se ha descrito en CCR hereditario no Lynch. Estos casos hereditarios forman parte del grupo hereditario sin poliposis (HNPCC) y con ausencia de alteración del sistema reparador (MSS). El 50% de las familias que cumplen los estrictos criterios clínicos de Ámsterdam (inicialmente ideados para identificar a familias con síndrome de Lynch) presentan este fenotipo. Los familiares afectados presentan CCR más tarde que los casos Lynch pero significativamente antes que los casos esporádicos, y el conjunto de cánceres extracolónicos asociados a este fenotipo es más reducido38. Los tumores MSS HNPCC, o tipo-X39, muestran un grado significativamente menor de metilación del gen LINE-1 (marcador de metilación global del genoma) que cualquier otro tipo de CCR, incluyendo tumores esporádicos (MSS y MSI), y casos con síndrome de Lynch40. Dado este bajo grado de metilación global, es posible que en el mecanismo carcinogénico de estos tumores tenga un papel fundamental la activación de oncogenes.
Dianas epigenéticas en el diseño de nuevos fármacosLa utilización de dianas epigenéticas para el desarrollo de nuevos tratamientos de quimioterapia está emergiendo como una estrategia con gran potencial dado que, en principio, las alteraciones epigenéticas son potencialmente reversibles. Así, el fármaco revertiría la represión génica de genes oncosupresores. Estos agentes modificarían los patrones de metilación del ADN o el estado de las histonas en las células tumorales a través de la alteración de la actividad de las enzimas responsables del establecimiento y mantenimiento de estos patrones. Entre estos fármacos, los más estudiados son los agentes demetilantes que funcionan inactivando las metiltransferasas del ADN (DNMT). Estas enzimas son responsables de transferir el grupo metilo al carbono 5 del nucleótido citosina. Las DNMT pueden regular la metilación de novo o mantener los patrones de metilación. En la actualidad, 2 inhibidores de DNMTs (azacitidine y decitabine) están aprobados por la administración norteamericana para su uso en pacientes con enfermedades hematológicas. Estudios clínicos están evaluando el uso de dosis bajas de inhibidores de DNMT en diferentes cánceres, incluyendo el de colon, para minimizar la toxicidad. En esta línea, zebularine es mucho menos tóxico y su solubilidad en agua permitiría su administración oral41. Experimentos in vitro en líneas celulares de CCR demuestran la reactivación de la expresión de genes hipermetilados en presencia de dosis bajas de decitabine y el inhibidor de acetilasas de histonas tricostatin A42.
Por último, estos fármacos pueden ayudar a mejorar la resistencia a otros agentes quimioterapéuticos reactivando genes como MLH1 y restableciendo así la acción de otros agentes como 5-fluorouracil en los tumores MSI43.
En general, aunque los efectos de estos agentes son prometedores, hay que ser cautelosos dada su falta de especificidad lo que puede resultar en la afectación del genoma en general y reactivar oncogenes, facilitando así la progresión del tumor. Afortunadamente, recientes estudios han demostrado que azacitidine y decitabine inducen la demetilación de genes específicos y no promueven una demetilación al azar44.
Efecto de la dieta en el cáncer colorrectal. Alteración de los patrones de metilación del ADN por parte de nutrientesLa incidencia de CCR es varias veces superior en EEUU, Australia, Nueva Zelanda, Europa o Japón con relación a las regiones menos desarrolladas de África y del sur y centro de Asia. Por otro lado, algunos estudios sugieren que el riesgo de desarrollar CCR aumenta rápidamente hasta asemejarse al de la población de acogida en inmigrantes procedentes de regiones con baja incidencia. Estos hechos evidencian la fuerte influencia de factores ambientales en el desarrollo de este cáncer.
En cuanto a la dieta, estudios epidemiológicos han sugerido que dietas ricas en fruta, vegetales y fibra y bajas en consumo de carne roja están asociadas a un menor riesgo de CCR. Sin embargo, en la mayoría de los casos estos efectos positivos no se han podido demostrar en estudios de intervención45. Las limitaciones de muchos de estos trabajos son debidas a su diseño. Así, los estudios de casos y controles tienden a presentar un mayor sesgo porque la información sobre la dieta se recoge de manera retrospectiva y los controles pueden no representar la subpoblación de la cual se han reclutado los casos. En los estudios aleatorizados, posiblemente el tiempo de intervención no es suficiente para poder evaluar el efecto de estos alimentos que incluyen distintos componentes. Por estos motivos, el papel de la dieta en el desarrollo del cáncer en la actualidad se dirige hacia el estudio de los efectos de componentes específicos de los alimentos. El estudio del mecanismo de acción de estos elementos puede resultar en el potencial uso de estos componentes como agentes quimiopreventivos. La quimioprevención utiliza agentes farmacológicos o naturales para impedir, disminuir o atrasar el progreso carcinogénico en su estado más temprano. Desde este punto de vista, se han identificado diferentes compuestos de la dieta que están relacionados con mecanismos clave para la metilación del ADN, como la alteración de la disponibilidad de donadores de los grupo metil y la alteración de la actividad de las enzimas involucradas en el proceso de metilación, especialmente las DNMT.
Componentes del metabolismo de un-carbono (folato, vitamina B)El primer mecanismo es el más estudiado y afecta principalmente al metabolismo de un-carbono y la regulación de SAM, el principal donador de metilos de la célula. Los nutrientes que pueden afectar este metabolismo incluyen el folato, las vitaminas B6 y B12, la colina y la metionina. Su carencia se ha asociado con el desarrollo de numerosos cánceres.
Un número significativo de estudios clínicos ha mostrado que existe una relación entre el nivel de folato y el riesgo de CCR, pero algunos datos resultan paradójicos. Así, un metanálisis de estudios prospectivos a gran escala ha mostrado un riesgo ligeramente reducido en pacientes que consumen una cantidad elevada de folato, aunque este efecto sería altamente dependiente de la cantidad, del tiempo de intervención y del estado de salud del individuo. Por otro lado, estudios recientes muestran que elevadas dosis de folato, presentes en suplementos vitamínicos, podrían tener un efecto potenciador del desarrollo de neoplasias colónicas46.
El folato es esencial para la síntesis de purinas y el nucleósido timidina. Si el nivel de folato es bajo, se produce un desequilibrio entre purinas y pirimidinas y se introducen alteraciones en la síntesis normal del ADN. Así la deficiencia de folato puede: a) inducir alteraciones en el ADN; b) comprometer su reparación, y c) modular el epigenoma.
Diferentes estudios han sugerido un efecto de hipometilación del genoma, pero también una hipermetilación de zonas específicas. De hecho, la depleción radical de folato en experimentos in vitro e in vivo se traduce en una disminución general y gen-específica de metilación. No obstante, el efecto en la metilación de citosinas debido únicamente a una deficiencia en el nivel de folato es un hallazgo que ha resultado ser menos reproducible. Posiblemente, el efecto del folato no sea causado mayoritariamente por efectos en la metilación del ADN y sea el resultado de una suma de efectos a distintos niveles.
PolifenolesLos polifenoles constituyen uno de los grupos más extensos de fitoquímicos. Sus funciones principales son: a) proteger las plantas del estrés fotosintético y de los compuestos reactivos del oxígeno, y b) prevenir su consumo por parte de los animales herbívoros. Los flavonoides y ácidos fenólicos son los principales compuestos polifenólicos en los alimentos.
El epigalocatequin galato, procedente del té verde, es capaz de reactivar genes silenciados por hipermetilación en líneas celulares de CCR. Por otro lado, la suplementación de líneas celulares de leucemia con cúrcuma, uno de los principales componentes del curry, induce una disminución global de la metilación del ADN comparable al efecto de decitabine. Estudios in vitro e in vivo sugieren que la cúrcuma también posee importantes efectos en otras alteraciones epigenéticas, ya que es un potente modificador de las histonas47.
Los efectos in vivo han resultado más difíciles de comprobar, aunque algunos trabajos recientes han confirmado el efecto protector de algunos polifenoles sobre el CCR. Así, la ingesta de 60g/d de frambuesas durante 9 semanas resultó en la demetilación de genes supresores relacionados con una de las vías principales del desarrollo del CCR y una disminución de la expresión de DNMT. Estos efectos son causados por el reveratrol, el polifenol que se encuentra en las bayas como uvas y tomates48.
SelenioEstudios de intervención han mostrado el efecto protector del selenio en varios cánceres, incluyendo el CCR46. El selenio es un elemento esencial con propiedades antioxidantes y proapoptóticas, que también se ha asociado con la metilación del ADN. Davis et al.49 mostraron que en colon e hígado, la deficiencia de selenio provoca una demetilación global y una metilación de p53 y p16, sugiriendo que el mecanismo de acción sería a través de la metilación del ADN.
Así pues, algunos nutrientes se están revelando como importantes agentes quimiopreventivos mediante su capacidad de revertir alteraciones epigenéticas. Estudios clínicos de intervención con un largo seguimiento se hacen imprescindibles para evaluar estos potenciales efectos antineoplásticos en las personas.
En resumen, los fenómenos epigenéticos están cobrando una significativa relevancia en los últimos tiempos y su implicación en el desarrollo del CCR tanto esporádico como hereditario se está haciendo evidente. Este hecho está facilitando el desarrollo de distintas aproximaciones diagnósticas y quimioterápicas basadas en el conocimiento de estas alteraciones, cuya utilización clínica veremos en un futuro muy próximo.
FinanciaciónBeca del Departamento de Medicina y Centro del Cáncer de la University of Illinois at Chicago, y de Sirazi Foundation.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.

