




La enfermedad de Wilson es una patología genética multiorgánica causada por la acumulación excesiva de cobre en el organismo. La afectación hepática es la inicial y principal, y puede suponer desde una hepatitis leve y transitoria, hasta el debut en forma de cirrosis o insuficiencia hepática aguda grave. En el seguimiento, hasta un 20-30% de estos pacientes evolucionan a cirrosis hepática. En la práctica clínica, la monitorización de la fibrosis hepática se realiza mayoritariamente mediante métodos indirectos y no invasivos (analíticas, elastografía hepática, ecografías) a semejanza de otras hepatopatías crónicas más prevalentes. No obstante, a pesar de que la elastografía constituye una herramienta de gran valor, la evidencia de su utilidad en Wilson es muy limitada. En esta revisión se repasa la información disponible y sus limitaciones para el seguimiento de la enfermedad de Wilson.
Wilson's disease is a sistemic genetic disease caused by the excessive accumulation of copper. The first and main involvement is in the liver, which can range from mild and transient elevation of transaminases to the onset of an overt cirrhosis or acute liver failure. It is known that up to 20-30% of these patients may evolve to liver cirrhosis during follow-up. In clinical practice, liver fibrosis is assessed mainly by using indirect and non-invasive tools (laboratory tests, liver elastography, ultrasound), similar to other prevalent chronic liver diseases. However, despite the fact that liver elastography is a valuable tool in general hepatology, the evidence of its usefulness and accuracy in Wilsońs disease is scarce. This review summarizes the available scientific data and their limitations in Wilson's disease.
La enfermedad de Wilson (EW) es un trastorno del metabolismo del cobre, de herencia autosómica recesiva, caracterizado por el depósito anormal de cobre en los tejidos, y con manifestaciones principalmente hepáticas, neuropsiquiátricas, corneales y hematológicas. La prevalencia poblacional estimada de la EW es globalmente baja (1/30.000), por lo que se la considera una enfermedad rara (https://www.orpha.net). No obstante, la prevalencia genética de portadores de alguna mutación patogénica en la población es aproximadamente de 1/90, sugiriendo que la enfermedad podría estar infradiagnosticada debido a una
penetrancia clínica variable1. La EW está causada por mutaciones en homozigosis o heterozigosis compuesta del gen ATP7B (cromosoma 13), que codifica para la proteína transportadora de cobre ATP7B2. Hasta la fecha se han descrito más de 1.000 mutaciones patogénicas distintas (HGMD Professional vs. 2020.2;), lo que refleja la gran complejidad genética de esta enfermedad.
La proteína ATP7B es ubicua, pero su principal expresión es a nivel de las células hepáticas. Su función es el secuestro del cobre intracelular para proveer el sustrato en la síntesis de la ceruloplasmina (principal proteína transportadora del cobre sérico), así como participar en la excreción biliar del cobre. Así, en condiciones fisiológicas de un individuo normal, el cobre sobrante no unido a ceruloplasmina será eliminado por las heces. La disfunción de este transportador ATP7B en la EW condiciona alteraciones en la síntesis de la ceruloplasmina (que es altamente inestable y degradada de forma rápida), así como imposibilidad para excretar adecuadamente el cobre por la bilis, lo que en última instancia lleva al acúmulo de cobre en dosis tóxicas3–5, con gran potencial de oxidación y daño celular en los tejidos6.
El órgano con alteraciones más frecuentes y tempranas en la EW es el hígado: la EW debe considerarse en el diagnóstico diferencial de cualquier hepatopatía, tanto en formas agudas como crónicas7. La forma de presentación hepática es muy heterogénea: desde la detección casual de hepatomegalia, hígado graso, alteraciones moderadas de las transaminasas o cirrosis, hasta casos de hepatitis aguda grave o fulminante. La edad de presentación también es muy variable; si bien la EW se ha considerado clásicamente una enfermedad de pacientes jóvenes (<40 años), algunos pacientes han sido diagnosticados en edades habitualmente impropias de la enfermedad (>70 años) sugiriendo la existencia de formas clínicas de menor penetrancia8,9. La presencia de alteraciones hepáticas asociadas a síntomas psiquiátricos y/o neurológicos, anemia hemolítica no inmune o los antecedentes en familiares de primer grado facilitan el diagnóstico o sospecha de EW. Los parámetros más usados para el diagnóstico son: niveles de ceruloplasmina sérica (típicamente baja ≤20mg/dl), cobre en orina de 24h (típicamente alta, ≥ 100mcg), y cobre en tejido hepático seco (típicamente elevado, ≥ 250mcg/g)7,10. No obstante, ninguno de estos parámetros es suficientemente potente para asegurar el diagnóstico de forma aislada, con S/Esp variables11. En los últimos años, se ha propuesto la medición directa del cobre intercambiable (CuEXC o fracción de cobre «libre» no unido a ceruloplasmina), como un marcador diagnóstico de gran potencia, si bien no se utiliza de forma generalizada (a excepción de Francia)12–14. En la práctica clínica, en especial en pacientes con presentación hepática y datos analíticos no concluyentes, se sugiere la combinación de diferentes parámetros clínicos y analíticos recogidos en los criterios de Leipzig de 200115 y avalados por las guías europeas de 20127. Actualmente, el único marcador con suficiente capacidad diagnóstica de EW es un estudio genético positivo. La histología hepática de la EW es inespecífica, pudiendo ser indistinguible de otros tipos de hepatopatía aguda o crónica2,16. Es frecuente observar esteatosis micro/macrovesicular. La única alteración patognomónica propia de la EW es el patrón de «hoja» de las crestas mitocondriales (microscopia electrónica), secundario al efecto tóxico del cobre sobre estas organelas16. La alteración en la integridad y función de las mitocondrias puede detectarse en estadios tempranos de la enfermedad pero no se realiza en la práctica clínica habitual17. La histoquímica para visualizar directa o indirectamente el exceso de cobre hepático (rodanina, orceína o tinción de plata) son técnicas con baja sensibilidad, especialmente en etapas iniciales, dada la distribución no homogénea del cobre hepático16,18.
Tras el diagnóstico de EW, el tratamiento está indicado de forma temprana, aún en ausencia de daño tisular demostrado, y está enfocado en conseguir un balance negativo del cobre para prevenir el desarrollo o progresión de las lesiones de órgano. El inicio temprano de tratamiento se asocia con un mejor pronóstico de los pacientes con EW7. Se basa en forzar la excreción urinaria de cobre por medio de agentes quelantes (D-penicilamina, trientina) o en evitar su absorción intestinal con sales de zinc7. En aquellos casos de fallo hepático agudo o cirrosis descompensada sin respuesta al tratamiento, está indicado el tratamiento definitivo mediante trasplante hepático2. En aquellos pacientes con manifestaciones predominantemente neurológicas, el tratamiento se basa en la combinación del tratamiento de la EW (priorizando dosis bajas de quelantes y /o sales de zinc) y el tratamiento sintomático. En relación con este último, se utilizan desde relajantes orales (benzodiacepinas, baclofén), intramusculares (toxina botulínica) o sistémicos (dantroleno), a agentes dopaminérgicos (levodopa, agonistas dopaminérgicos), así como neurolépticos ya sea para las manifestaciones hipercinéticas o las psiquiátricas, pero optando siempre por los neurolépticos por menor riesgo de efectos adversos extrapiramidales. En casos de distonía generalizada grave o tormenta distónica así como temblor refractario se puede considerar la estimulación cerebral profunda del globo pálido interno. Aquellos síntomas neurológicos que no mejoran tras 12 meses de tratamiento se deben considerar irreversibles. Recientemente se ha propuesto el trasplante hepático como alternativa de tratamiento de la EW con presentación neurológica refractaria19.
Seguimiento de la hepatopatía en pacientes con enfermedad de WilsonA pesar de ser una enfermedad descrita hace muchos años (1912)20, la EW sigue constituyendo una enfermedad rara con enormes retos en el diagnóstico, seguimiento y monitorización del tratamiento7,21,22. En lo que respecta a las manifestaciones hepáticas, a día de hoy, no existe estandarización ni consenso en el seguimiento prospectivo a medio/largo plazo de los pacientes con EW, por lo que cada centro actúa de acuerdo a su experiencia previa y en ocasiones se adaptan protocolos validados para otras hepatopatías más prevalentes. Generalmente los pacientes con EW se siguen en las consultas de Hepatología cada 6-12 meses. Las series publicadas de pacientes con EW estiman que en torno a un 20-30% de los pacientes evolucionarán a cirrosis hepática a lo largo del tiempo7,22. Los factores asociados a dicha progresión no están bien establecidos, pero podría asumirse que la falta de adherencia a la medicación, la realización de un tratamiento subóptimo o la presencia de comorbilidades, podrían influir en esta evolución desfavorable.
Seguimiento terapéutico y confirmación de la adherenciaUna de las principales dificultades en el seguimiento de los pacientes con EW pasa por la monitorización de la adherencia terapéutica. Como en otras enfermedades crónicas, la adherencia a los tratamientos a largo plazo es difícil de mantener, más aún en enfermedades con diagnósticos en edad pediátrica y/o juvenil (WHO report:https://www.who.int/home). La posología de los fármacos indicados para EW no es sencilla (varias tomas al día, necesidad de ayunas para garantizar absorción), hay con frecuencia problemas de seguridad y esto hace que la adherencia constituya un problema en muchos casos. No obstante, es sabido que una adherencia adecuada es clave para evitar la progresión de la enfermedad23,24. La falta de adherencia puede provocar empeoramientos rápidos de la enfermedad con consecuencias graves. La monitorización de la adherencia es compleja, pero pueden usarse los valores de zinc y cobre en orina de 24h. Así, en los pacientes en tratamiento con sales de zinc, una zincuria inferior a 2g o niveles de cobre de>100mcg/24h sugerirían mala adherencia; entre pacientes en tratamiento con quelantes, la cupruria debería mantenerse entre 200-500mcg/24h. Cifras mucho más altas o fluctuantes (fuera de la fase inicial de tratamiento) sugerirían deficiencias en la adherencia10 y mal control por tanto de los niveles del cobre sistémico.
El objetivo teórico de un buen tratamiento en la EW es alcanzar la remisión clínica (reversibilidad de síntomas/signos previos) y la respuesta bioquímica (normalización de las transaminasas). No obstante, estos objetivos no suelen ocurrir antes de los 6-12 meses de tratamiento, y pueden ser más tardíos en pacientes con clínica neurológica. Las guías clínicas7,21,25 de adultos y edad pediátrica recomiendan disminuir las dosis de los fármacos una vez alcanzada la respuesta clínica y bioquímica, para no inducir una depleción excesiva de cobre con efectos iatrogénicos. Algunos pacientes, a pesar del tratamiento, pueden no obstante, no normalizar totalmente las transaminasas durante el seguimiento2. No existe consenso claro acerca de cuál debe ser el momento y el algoritmo de descenso de la medicación en la fase de mantenimiento de la EW, pero las fichas técnicas de los fármacos sugieren no exceder los 450mg de acetato de zinc, los 750mg de D-penicilamina o los 1250mg de trientina al día (fichas técnicas: https://cima.aemps.es/cima/publico/home.html).
Desde el punto de vista bioquímico, el seguimiento de los pacientes está dirigido a garantizar que los tratamientos mantengan un equilibrio adecuado en la homeostasis del cobre: a saber, niveles de cobre en orina 24h <75-100μg (en pacientes tratados con zinc) o entre 200–500μg/día (en pacientes en tratamiento con quelantes), o unos niveles de «cobre libre» inferiores a 50-75μg/l (en todos). El cálculo del cobre libre (NCC, de non-ceruloplasmin bound copper por sus cifras en inglés) es un cálculo indirecto realizado a partir de los niveles de ceruloplasmina (NCC=cobre total[μg/dl]–3,15 x ceruloplasmina[mg/dl]); el NCC es un marcador bioquímico de cálculo complejo, de gran variabilidad, y con resultados no valorables o incluso negativos hasta en el 25% de los pacientes10. Conociendo sus limitaciones, podría ser complementario al resto de los marcadores, pero no un marcador único que guíe nuestro seguimiento clínico.
En los últimos años, se ha propuesto la medición del cobre intercambiable (CuEXC) como un marcador de interés12,13. Esta fracción representaría el cobre circulante libre, el cobre unido a otras proteínas transportadoras minoritarias (albúmina, transcupreína) o aquel unido de forma lábil a la ceruloplasmina, (y fácilmente intercambiable en presencia de quelantes como el EDTA), y que tendria potencial para generar toxicidad. Este marcador experimental ha demostrado ser útil en el diagnóstico de la EW, así como en la diferenciación fenotípica de pacientes sintomáticos, presintomáticos y portadores13. Los niveles de CuEXC son mayores en pacientes con enfermedad extrahepática que en pacientes con enfermedad hepática aislada14. Sin embargo, su utilidad en el seguimiento de los pacientes con EW no ha sido validado hasta la fecha10.
Así pues, la monitorización bioquímica durante el seguimiento se basa actualmente en la combinación de cobre en orina de 24h, transaminasas y eventualmente NCC, y debería ser útil para:
- a)
Determinar con fiabilidad si un paciente es adherente.
- b)
Evaluar si la dosis de fármaco es la adecuada, y ajustarla convenientemente.
- c)
En caso de adherencia y dosis adecuada, valorar modificar el tratamiento si los objetivos bioquímicos y clínicos no se alcanzan.
En un paciente con EW, uno de los objetivos del tratamiento pasa por la normalización de las transaminasas. No obstante, dicho objetivo bioquímico no suele ocurrir antes de los 6-12 meses de iniciado el tratamiento. Es importante recordar que en pacientes en los que existe afectación hepática, los quelantes deberían constituir la primera línea de tratamiento26. La alteración de las transaminasas una vez normalizadas bajo tratamiento debe instarnos a estudiar si la dosis y adherencia a la terapia en nuestro paciente sigue siendo adecuada, o pueden haberse sumado comorbilidades o noxas hepáticas a la EW. La misma recomendación aplicaría para aquellos pacientes en los que las transaminasas no estaban alteradas de inicio y en los que observamos cambios durante el seguimiento.
No existe, sin embargo, correlación significativa entre los parámetros analíticos (transaminasas) y la gravedad de la enfermedad27,28. Un ejemplo típico lo constituiría el fallo hepático agudo por EW, que se encuentra frecuentemente asociado a niveles solo moderadamente elevados de transaminasas2, que contrastan con el incremento marcado de la bilirrubina y una coagulopatía severa, asociadas o no a anemia hemolítica Coombs negativa. En pacientes con cirrosis hepática o insuficiencia hepática, la monitorización de la función hepatocelular es fundamental. El índice pronóstico de EW modificado de Dhawan29 se basa en marcadores de función hepática como albúmina, AST, INR, bilirrubina y leucocitos; un score> 11 puntos se relaciona con alta probabilidad de fallecimiento a corto plazo y es indicación para trasplante hepático. En aquellos pacientes que no tengan indicación de trasplante hepático y en los que esté indicado el tratamiento médico se deben realizar diversas medidas: tratamiento con quelantes de cobre, restricción dietética, evitar hepatotóxicos y realizar el despistaje de posibles complicaciones derivadas de hipertensión portal (varices esofágicas y/o hepatocarcinoma).
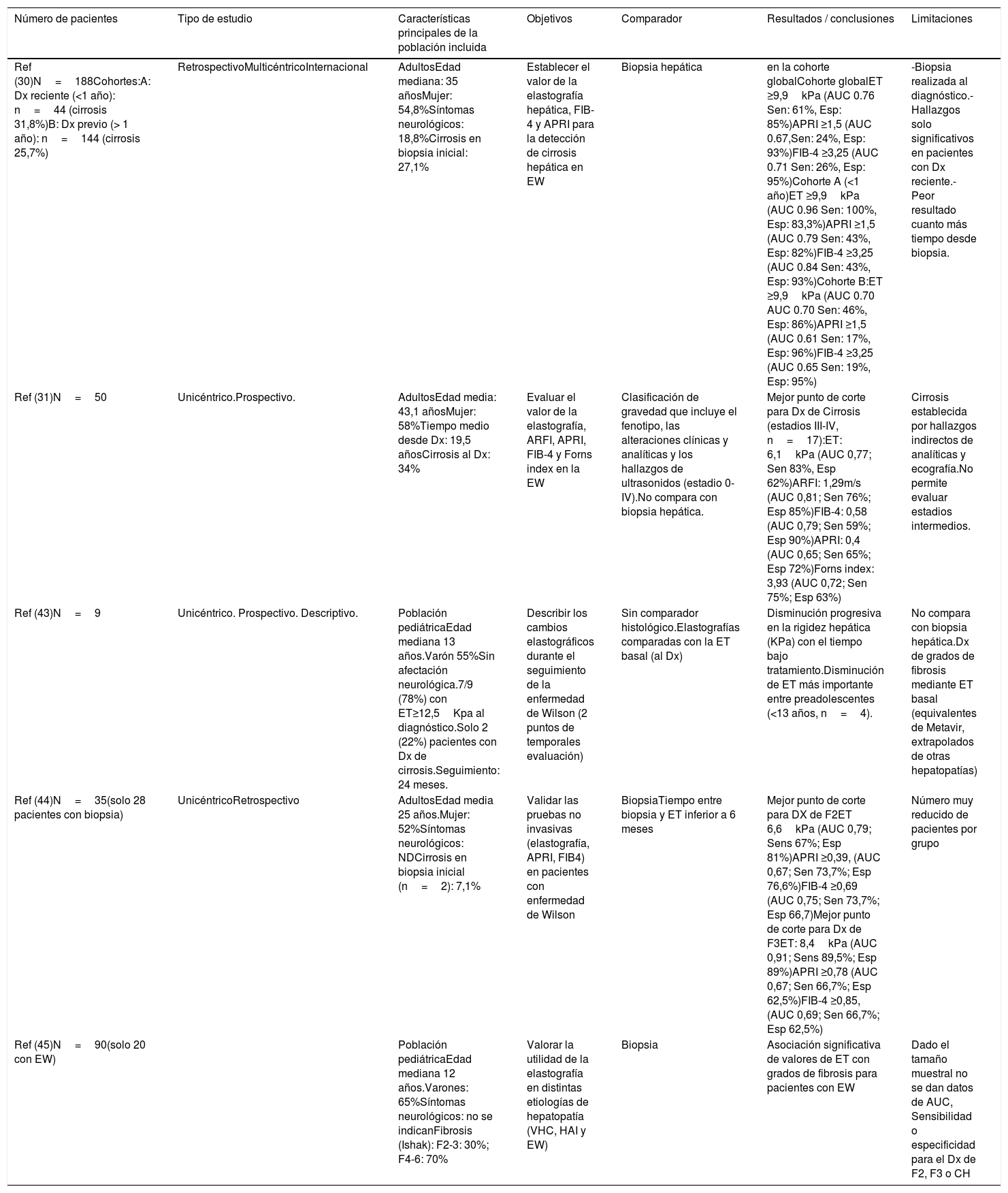
Evaluación no invasiva de la fibrosis hepáticaDesde hace una década, el desarrollo de los métodos no invasivos y su validación en las enfermedades hepáticas crónicas ha permitido reducir el número de procedimientos invasivos para el seguimiento de los pacientes. Un ejemplo es la elastografía hepática (ET) o fibroscan, que estima la fibrosis hepática mediante la detección de la velocidad de propagación de ondas30,31. El fibroscan ha sido validado para la monitorización de la fibrosis y el diagnóstico de cirrosis en la mayoría de las hepatopatías prevalentes: hepatitis C:12,5-14kPa32, hepatitis B: 12.4kPa28,33 o enfermedad por hígado graso no alcohólico: 8,7kPa32. Sin embargo, el uso de la elastografía hepática en la EW es más limitado y hay pocos estudios longitudinales al respecto, en consonancia con la baja prevalencia de la enfermedad (resumidos en la tabla 1). Un reciente estudio multicéntrico centroeuropeo34 evaluó el valor de la elastografía hepática en 188 pacientes con EW y biopsia hepática al diagnóstico (o como estudio en explante). En este trabajo, el punto de corte de 9,9kPa fue capaz de detectar cirrosis con precisión (S: 100%, Esp: 83%) entre aquellos pacientes con diagnóstico reciente (seguimiento <1 año) (n=44, 23%). Sin embargo, en los 144 pacientes con mayor tiempo de seguimiento (entre 1-10 años desde el diagnóstico al fibroscan), su precisión fue significativamente inferior (S: 46% Esp: 86%). Otros estudios con tamaño muestral más reducido31, y heterogeneidad en el comparador gold standard, han intentado validar la ET como método de seguimiento en los pacientes con EW, sin que hayan sido capaces de establecerse puntos de corte óptimos para la monitorización de la progresión de la fibrosis. Si bien es cierto que el fibroscan parece ser mayor cuanto mayor es el grado de fibrosis hepática también en estos pacientes, la tendencia es a observar valores inferiores a los observados en otras hepatopatías crónicas.
Revisión de estudios con evaluación no invasiva de la fibrosis hepática en la EW
| Número de pacientes | Tipo de estudio | Características principales de la población incluida | Objetivos | Comparador | Resultados / conclusiones | Limitaciones |
|---|---|---|---|---|---|---|
| Ref (30)N=188Cohortes:A: Dx reciente (<1 año): n=44 (cirrosis 31,8%)B: Dx previo (> 1 año): n=144 (cirrosis 25,7%) | RetrospectivoMulticéntricoInternacional | AdultosEdad mediana: 35 añosMujer: 54,8%Síntomas neurológicos: 18,8%Cirrosis en biopsia inicial: 27,1% | Establecer el valor de la elastografía hepática, FIB-4 y APRI para la detección de cirrosis hepática en EW | Biopsia hepática | en la cohorte globalCohorte globalET ≥9,9kPa (AUC 0.76 Sen: 61%, Esp: 85%)APRI ≥1,5 (AUC 0.67,Sen: 24%, Esp: 93%)FIB-4 ≥3,25 (AUC 0.71 Sen: 26%, Esp: 95%)Cohorte A (<1 año)ET ≥9,9kPa (AUC 0.96 Sen: 100%, Esp: 83,3%)APRI ≥1,5 (AUC 0.79 Sen: 43%, Esp: 82%)FIB-4 ≥3,25 (AUC 0.84 Sen: 43%, Esp: 93%)Cohorte B:ET ≥9,9kPa (AUC 0.70 AUC 0.70 Sen: 46%, Esp: 86%)APRI ≥1,5 (AUC 0.61 Sen: 17%, Esp: 96%)FIB-4 ≥3,25 (AUC 0.65 Sen: 19%, Esp: 95%) | -Biopsia realizada al diagnóstico.- Hallazgos solo significativos en pacientes con Dx reciente.- Peor resultado cuanto más tiempo desde biopsia. |
| Ref (31)N=50 | Unicéntrico.Prospectivo. | AdultosEdad media: 43,1 añosMujer: 58%Tiempo medio desde Dx: 19,5 añosCirrosis al Dx: 34% | Evaluar el valor de la elastografía, ARFI, APRI, FIB-4 y Forns index en la EW | Clasificación de gravedad que incluye el fenotipo, las alteraciones clínicas y analíticas y los hallazgos de ultrasonidos (estadio 0-IV).No compara con biopsia hepática. | Mejor punto de corte para Dx de Cirrosis (estadios III-IV, n=17):ET: 6,1kPa (AUC 0,77; Sen 83%, Esp 62%)ARFI: 1,29m/s (AUC 0,81; Sen 76%; Esp 85%)FIB-4: 0,58 (AUC 0,79; Sen 59%; Esp 90%)APRI: 0,4 (AUC 0,65; Sen 65%; Esp 72%)Forns index: 3,93 (AUC 0,72; Sen 75%; Esp 63%) | Cirrosis establecida por hallazgos indirectos de analíticas y ecografía.No permite evaluar estadios intermedios. |
| Ref (43)N=9 | Unicéntrico. Prospectivo. Descriptivo. | Población pediátricaEdad mediana 13 años.Varón 55%Sin afectación neurológica.7/9 (78%) con ET≥12,5Kpa al diagnóstico.Solo 2 (22%) pacientes con Dx de cirrosis.Seguimiento: 24 meses. | Describir los cambios elastográficos durante el seguimiento de la enfermedad de Wilson (2 puntos de temporales evaluación) | Sin comparador histológico.Elastografías comparadas con la ET basal (al Dx) | Disminución progresiva en la rigidez hepática (KPa) con el tiempo bajo tratamiento.Disminución de ET más importante entre preadolescentes (<13 años, n=4). | No compara con biopsia hepática.Dx de grados de fibrosis mediante ET basal (equivalentes de Metavir, extrapolados de otras hepatopatías) |
| Ref (44)N=35(solo 28 pacientes con biopsia) | UnicéntricoRetrospectivo | AdultosEdad media 25 años.Mujer: 52%Síntomas neurológicos: NDCirrosis en biopsia inicial (n=2): 7,1% | Validar las pruebas no invasivas (elastografía, APRI, FIB4) en pacientes con enfermedad de Wilson | BiopsiaTiempo entre biopsia y ET inferior a 6 meses | Mejor punto de corte para DX de F2ET 6,6kPa (AUC 0,79; Sens 67%; Esp 81%)APRI ≥0,39, (AUC 0,67; Sen 73,7%; Esp 76,6%)FIB-4 ≥0,69 (AUC 0,75; Sen 73,7%; Esp 66,7)Mejor punto de corte para Dx de F3ET: 8,4kPa (AUC 0,91; Sens 89,5%; Esp 89%)APRI ≥0,78 (AUC 0,67; Sen 66,7%; Esp 62,5%)FIB-4 ≥0,85, (AUC 0,69; Sen 66,7%; Esp 62,5%) | Número muy reducido de pacientes por grupo |
| Ref (45)N=90(solo 20 con EW) | Población pediátricaEdad mediana 12 años.Varones: 65%Síntomas neurológicos: no se indicanFibrosis (Ishak): F2-3: 30%; F4-6: 70% | Valorar la utilidad de la elastografía en distintas etiologías de hepatopatía (VHC, HAI y EW) | Biopsia | Asociación significativa de valores de ET con grados de fibrosis para pacientes con EW | Dado el tamaño muestral no se dan datos de AUC, Sensibilidad o especificidad para el Dx de F2, F3 o CH |
AUC: área bajo la curva; CH: cirrosis hepática; DX: diagnóstico; Esp: especificidad; ET: elastografía de transición; EW: enfermedad de Wilson; F: grado de fibrosis; HAI; enfermedad autoinmune; Ref: referencia; Sen: sensibilidad; VHC: virus de la hepatitis C.
Otros marcadores serológicos desarrollados para evaluar la fibrosis hepática de forma no invasiva (como el APRI, FIB-4, índice de Forns) han demostrado su utilidad en otras hepatopatías para descartar la presencia de cirrosis, debido a su alta especificidad, con puntos de corte de 1,5 y 3,25 para APRI y FIB-4 respectivamente32. Sin embargo, la utilidad de estos marcadores no invasivos de fibrosis en pacientes con EW no ha sido tampoco establecida con fiabilidad hasta la fecha. En el estudio de Paternostro et al.34, se describe la asociación de APRI y FIB-4 con cirrosis histológica, pero solo en aquellos pacientes con diagnóstico reciente (< 1 año), en los que la elastografía ya detectaba adecuadamente la cirrosis. Otros estudios35 describen la asociación entre estos marcadores y la elastografía, pero sin que exista aproximación comparativa mediante biopsia hepática, por lo que queda limitado el valor de estas observaciones.
Evaluación invasiva de la fibrosis hepáticaLa cuantificación directa de la fibrosis hepática se ha realizado clásicamente mediante biopsia hepática, que constituye el estándar oro de comparación. La biopsia hepática, de acceso transparietohepático o transyugular, es un procedimiento invasivo no exento de riesgos, en el que se obtiene un fragmento de parénquima hepático por punción36. En los últimos años, el desarrollo de estrategias no invasivas ha reducido en gran medida la necesidad de realizar biopsias hepáticas en la mayoría de los pacientes.
Si bien la biopsia hepática está firmemente aceptada como parte del algoritmo diagnóstico en la EW tanto en edad pediátrica como adulta7,21,25,37, la utilidad de las biopsias de seguimiento es menor. Como ha ocurrido con muchas otras hepatopatías crónicas, la invasividad inherente a la biopsia ha hecho que quede restringida a casos con empeoramiento clínico inexplicable o casos con dudas diagnósticas. Sin embargo, en el caso de la EW, teniendo en cuenta la ausencia de especificidad de la afectación histológica, y las limitaciones de las técnicas no invasivas mencionadas, las biopsias de seguimiento en la EW podrían tener varios objetivos:
- a)
Cuantificar el cobre en tejido tras un tiempo variable de tratamiento.
- b)
Estadiar la enfermedad y evaluar la progresión histológica bajo tratamiento.
- c)
Descartar la coexistencia de otras hepatopatías añadidas.
En lo que respecta al valor de la cuantificación de cobre intrahepático en pacientes con EW bajo tratamiento, la evidencia disponible es limitada y en muchos casos antigua. Gibbs et al.38 reportaron en 1990 una asociación entre el tratamiento con tetratiomolibdato y la disminución hepática de cobre en tejido, mientras que otros estudios más recientes de series pequeñas seguidas histológicamente en el tiempo27,39 no fueron capaces de detectar asociación entre los niveles de cobre intrahepático y la progresión o no de la enfermedad histológica, o el tipo de tratamiento recibido. Algunas explicaciones a la falta de normalización del cobre o la falta de asociación de los niveles intrahepáticos con la mejoría o empeoramiento histológico, podrían ser la gran heterogeneidad del depósito de cobre en el parénquima, sumado a la variabilidad de la muestra, el tiempo variable de seguimiento y a que algunos tratamientos (especialmente las sales de zinc) podrían llevar a la acumulación de formas no tóxicas de cobre, que quedarían igualmente depositadas en el hígado. No obstante, el pequeño tamaño muestral de estos estudios que evalúan longitudinalmente la EW mediante biopsia hace que sea complicado poder sacar conclusiones al respecto35.
La evaluación de la fibrosis hepática mediante biopsia a lo largo del tiempo en pacientes con EW se ha evaluado en pocos estudios, con resultados heterogéneos y con descripciones histopatológicas variables. Cope-Yokoyama et al27. incluyeron 12 pacientes con EW seguidos durante una media de 5 años (rango 1-12 años), en los que realizaban biopsias percutáneas de seguimiento (al menos dos, una al diagnóstico y otra tras una media de tiempo de 4 años). La mayoría de los casos (n=11) fueron tratados con el mismo tratamiento durante todo el tiempo del estudio. Tras el examen ciego de las biopsias por parte de 2 patólogos con experiencia, los autores reportaron estabilidad o mejoría de las lesiones en 6 casos (mejoría de la esteatosis en 3, mejoría de la fibrosis en 1), mientras que observaron progresión de las lesiones histológicas (inflamación y/o fibrosis) en el resto (n=6). Con estos datos, los autores reportaron una tasa de fibrosis de 0 al año durante los primeros 4 años (entre la primera y segunda biopsia), que aumentaba a 0,25 unidades de fibrosis en 3 años entre la segunda y tercera biopsia (realizada a solo 4 pacientes), sin que pudieran determinarse los factores predictivos de progresión histológica. Dados los resultados, los autores sugerían la realización de biopsia hepática a los 3 años como aproximación asistencial justificable, aun a falta de estudios longitudinales con mayor tamaño muestral. Otros estudios que han evaluado la progresión histológica de los pacientes con EW40–45 y que incluyen conjuntamente a 42 pacientes, muestran resultados variables, con mejoría global en los parámetros de fibrosis, inflamación y esteatosis a lo largo del tiempo, en biopsias separadas entre 2-10 años. En conjunto, podríamos afirmar que actualmente no hay evidencia suficiente para recomendar la biopsia hepática de seguimiento, especialmente en aquellos pacientes con estabilidad analítica, terapéutica y clínica. No obstante, las limitaciones de todas estas formas de seguimiento no invasivas son indiscutibles, por lo que la biopsia hepática no debería descartarse a lo largo del tiempo.
Por último, hay que tener en cuenta que los pacientes con EW pueden tener a lo largo de la vida otros problemas hepáticos. La coexistencia en estos pacientes de un síndrome metabólico no es infrecuente, y el consumo de alcohol puede ser relevante. La biopsia hepática podría ayudar a detectar rasgos histológicos de otras comorbilidades y servir de ayuda para el ajuste de los tratamientos.
Seguimiento neurológico de los pacientes con enfermedad de WilsonTeniendo en cuenta que las manifestaciones neurológicas en la EW pueden ir desde alteraciones sutiles e intermitentes a formas agudas y extremadamente graves, es aconsejable que todo paciente con EW sea valorado por Neurología.
En los casos asintomáticos o paucisintomáticos se puede realizar seguimiento ambulatorio electivo basado en la historia y el examen clínico, siendo aconsejable realizar una resonancia magnética cerebral inicial, con posterior seguimiento de imagen en función de la evolución clínica y el seguimiento metabólico.
En los casos sintomáticos, aparte del tratamiento metabólico (quelantes, sales de zinc), hay que emplear los tratamientos sintomáticos que corresponda dependiendo de las manifestaciones neurológicas y se monitorizarán de forma más estrecha con resonancia magnética, que puede mostrar desde lesiones reversibles (signo del ojo panda en el mesencéfalo, alteraciones de señal en núcleos lenticulares) hasta daño irreversible (lesiones necrótico-quísticas en ganglios basales, tronco del encéfalo, cerebelo). Un estudio reciente ha propuesto los niveles plasmáticos de neurofilamento como biomarcador de afectación neurológica en la EW lo que podría incluir utilidad potencial en el seguimiento de la terapia quelante 46.
ConclusionesLa EW es una enfermedad rara con grandes retos actuales para el seguimiento clínico. La escasa literatura, las limitaciones de las técnicas no invasivas para la monitorización de la fibrosis, la dificultad para monitorizar el tratamiento y la adherencia por marcadores imperfectos, la escasez de opciones terapéuticas, o la coexistencia de otras enfermedades hepáticas, hacen difícil tratar de forma óptima a muchos de estos pacientes. En este contexto, la biopsia hepática podría seguir constituyendo una herramienta de utilidad tanto en el diagnóstico como en el seguimiento, especialmente en aquellos casos con evolución atípica o variable.
Conflicto de interesesZM es consultora de Alexion®, Orphalan®, DeepGenomics®. YC es consultor de Alexion®. GHQ, AD y XF declaran no tener ningún conflicto de intereses.

