




Las mielitis longitudinales extensas (LETM) fueron clásicamente relacionadas con los trastornos del espectro de la neuromielitis óptica (NMOSD) tanto definidas como limitadas. Sin embargo, los diagnósticos diferenciales incluyen una amplia gama de etiologías.
ObjetivoComunicar los diagnósticos diferenciales y el pronóstico de LETM observados en un grupo de pacientes en Buenos Aires, Argentina.
Pacientes y métodosEstudio multicéntrico retrospectivo transversal realizado en 2 hospitales de Buenos Aires desde junio del 2008 hasta junio del 2014. Criterios de inclusión: síndrome medular asociado a una lesión en la médula espinal con una extensión de 3 o más segmentos vertebrales contiguos en la resonancia magnética (RM). Datos bioquímicos, radiológicos y clínicos fueron evaluados. Asimismo, se aplicó la escala de discapacidad funcional de Winer-Hughes (WHFDS) a los 3 meses.
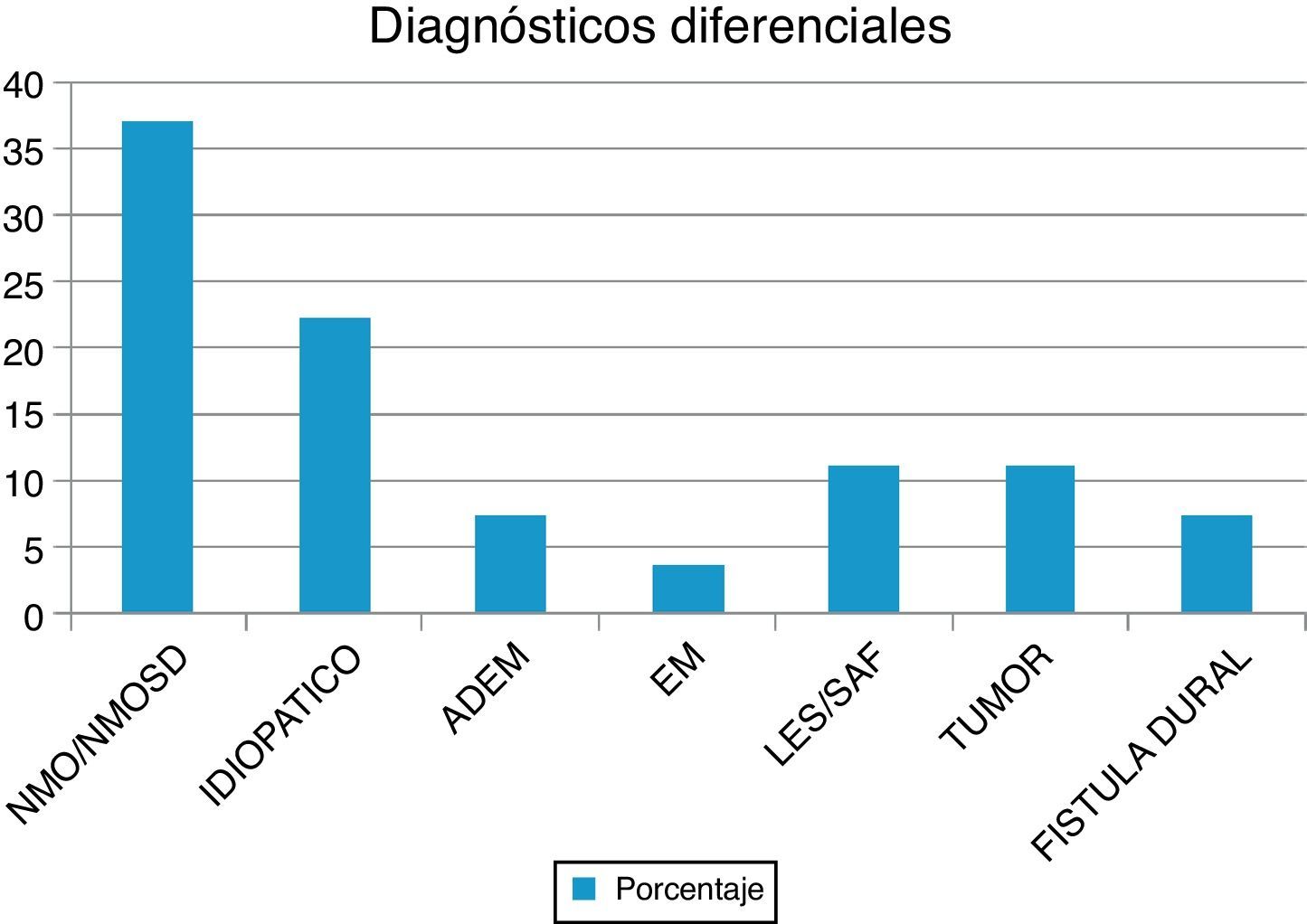
ResultadosSe evaluó a 27 pacientes, el 74% mujeres, edad (media): 35,22 años. NMO-IgG se realizó en el 66,6% y las bandas oligoclonales en el 71%. NMO-IgG se observó exclusivamente en pacientes con NMOSD (75%). La RM de encéfalo fue normal en el 59,2% y la media de segmentos afectados en RM espinal fue 7,9. Los diagnósticos diferenciales encontrados fueron: NMOSD (37%), idiopática (22,2%), lupus (11,1%), tumores (11,1%), fístula dural (7,4%), encefalomielitis diseminada aguda (7,4%) y esclerosis múltiple (3,7%). Los pacientes con ≥ 7 segmentos afectados tenían peor WHFDS (p < 0,001) y se asoció a etiología tumoral, vascular, lupus e idiopática.
ConclusionesEn nuestra cohorte, NMOSD seguidos por idiopática, fueron las causas más frecuentes de LETM. Las LETM tumorales, vasculares, lupus e idiopáticas pueden representar un grupo crítico con diferente pronóstico y tratamiento.
Longitudinally extensive myelitis (LETM) has classically been grouped with the full or limited neuromyelitis optica spectrum disorders (NMOSD). However, differential diagnosis reveals a wide range of aetiologies.
ObjectiveTo report on differential diagnosis and prognosis for LETM observed in a group of patients in Buenos Aires, Argentina.
Patients and methodsCross-sectional and retrospective multicentre study in two hospitals in Buenos Aires from June 2008 to June 2014. Inclusion criteria: medullary syndrome associated with spinal cord lesion extending for 3 or more contiguous spinal segments in magnetic resonance imaging (MRI). Clinical, radiological, and biochemical data were collected and subjects were rated on the Hughes functional disability scale (WHFDS) at 3 months.
ResultsWe evaluated 27 patients, 74% of whom were women; mean age was 35.22 years. The NMO-IgG antibody test was performed in 66.6% and oligoclonal band testing in 71%. NMO-IgG seropositivity was found exclusively in NMOSD patients (75%). Brain MRI was normal in 59.2% and revealed a mean of 7.9 affected spinal segments. Differential diagnoses revealed NMOSD (37%), idiopathic LETM (22.2%), lupus (11.1%), tumour (11.1%), dural fistula (7.4%), acute disseminated encephalomyelitis (7.4%), and a single case of multiple sclerosis (3.7%). Patients with lesions to ≥ 7 spinal segments showed poor recovery at 3 months (P<.001); these cases were associated with neoplastic, vascular, idiopathic, and lupus-related aetiologies.
ConclusionsThe most frequent causes of LETM in our cohort were NMOSD followed by idiopathic cases. Neoplastic, vascular, lupus-related, and idiopathic LETM may constitute a critical group with a distinct prognosis and other treatment needs.
La mielitis transversa longitudinal extensa (LETM) es definida como una lesión hiperintensa en T2 que afecta al menos 3 segmentos medulares en cortes sagitales de resonancia magnética (RM)1-3. Si bien la LETM es rara, es importante tener en cuenta que dicha patología se asocia a consecuencias clínicas severas, por lo que, una evaluación sistematizada para el reconocimiento etiológico y el tratamiento precoz son de suma importancia no solo para el pronóstico funcional a corto y largo plazo, sino también para prevenir ataques futuros en la médula espinal y/o en el sistema nervioso central en aquellas causas inflamatorias-desmielinizantes3-5. En la actualidad, a la LETM se la relaciona principalmente con la neuromielitis óptica (NMO) y su espectro (NMOSD)1-6. Sin embargo, existen múltiples diagnósticos diferenciales a tener en cuenta: enfermedades inflamatorias autoinmunes no NMO (encefalomielitis diseminada aguda [ADEM], esclerosis múltiple [EM], etc.), enfermedades autoinmunes sistémicas (lupus eritematoso sistémico [LES], síndrome de Sjögren, sarcoidosis, etc.), infecciosas y parainfecciosas (virus, bacterias, parásitos), paraneoplásicas (principalmente secundario al anticuerpo CRMP-5), neoplásicas (astrocitoma, ependimoma, metástasis [MTS] intramedulares, etc.), metabólicas (déficits de vitamina B12 o cobre), vasculares (infarto o fístulas durales), entre otras3. En el 2002, fueron propuestos los criterios diagnósticos para mielitis idiopática por The Transverse Myelitis Consortium Working Group para aquellos pacientes en que la etiología no esté aclarada aún1,2.
Debido a la falta de datos en nuestra región, nuestro objetivo es evaluar el diagnóstico etiológico, las características clínicas, paraclínicas y el pronóstico funcional a los 3 meses en una cohorte de pacientes con LETM en Buenos Aires, Argentina.
Pacientes y métodosEstudio retrospectivo descriptivo y transversal multicéntrico, donde se incluyó a pacientes atendidos en 2 hospitales públicos de la Ciudad de Buenos Aires (Hospital Carlos G. Durand y Hospital Teodoro Álvarez) mediante la revisión de historias clínicas, entre junio del 2008 y mayo del 2014. Se incluyó a todos los pacientes que cumplan con los criterios de inclusión (véase la tabla 1) y se evaluaron datos demográficos, clínicos, paraclínicos, radiológicos, etiológicos y la evolución funcional mediante la escala de discapacidad funcional de Winer-Hughes (WHFDS) a los 3 meses7. Respecto de los datos demográficos, se tuvieron en cuenta la edad de comienzo y el sexo. Además, se objetivó la presencia, en el momento de la evaluación neurológica, de trastornos motores, sensitivos y/o autonómicos. Los estudios inmunológicos y procoagulantes, así como la búsqueda de etiología infecciosa en suero, fueron realizados ante la sospecha diagnóstica según los criterios internacionales validados para cada etiología1,4,8-15. Asimismo, cabe aclarar que los anticuerpos antiacuaporina 4 (AQP4) (IgG-NMO) en suero fueron realizados mediante inmunofluorescencia indirecta confocal en cerebelo de mono. Las bandas oligoclonales (BOC) en líquido cefalorraquídeo (LCR) fueron analizadas mediante isoelectroenfoque. Los estudios de PCR específicos solo se realizaron ante la sospecha clínica y epidemiológica. La RM de cerebro y espinal fueron realizadas en equipos de 1.5 Tesla en toda la muestra y en el mismo equipo. Todos los pacientes fueron evaluados con RM de cerebro y columna al menos cervical y dorsal con y sin gadolinio, según criterio del médico a cargo se realizó además RM lumbar. Los hallazgos de las RM de encéfalo fueron clasificados en lesiones inespecíficas de sustancia blanca, lesiones que cumplan criterios de Swanton et al.12,13 y Montalban et al.14 para diseminación en espacio y tiempo (criterios de McDonald 201011); y típicas de NMO (hipotálamo, tálamo, tronco encefálico o cerebelo). A todos los pacientes se los siguió al menos 3 meses con exámenes neurológicos periódicos y se utilizó la WHFDS (0: saludable o recuperación completa; i: signos o síntomas menores que no limitan la actividad física; ii: capaz de caminar 5 m sin asistencia, andador o bastón, pero incapaz para trabajo manual, compras o trabajo doméstico; iii: capaz de caminar 5 m con asistencia, andador o bastón, iv: confinado a silla de ruedas o cama; v: requerimiento de ventilación asistida por lo menos una parte del día o de la noche, y vi: muerte) a los 90 días. Dicha escala fue categorizada en 2 grupos ≤ 2 y ≥ 3 puntos. Luego se comparó cantidad de segmentos espinales afectados vs. necesidad de ayuda para la marcha o silla de ruedas (≤ 2 vs. ≥ 3).
Criterios de inclusión
| Edad inicio: mayor a 18 años |
| Diagnóstico clínico de síndrome medular realizado por un neurólogo |
| Presentación clínica con déficit motor, sensitivo o trastornos esfinterianos atribuible a una lesión espinal en RM que afecte 3 o más segmentos |
| Progresión máxima del déficit hasta 21 días |
| Exclusión de etiología extraaxial por imágenes |
| Exclusión de traumatismo raquimedular |
| RM cerebro y médula (al menos cervicodorsal) con gadolinio realizado en el mismo equipo de estudio (1.5 Tesla) |
| Seguimiento por al menos 3 meses con escala de Winer-Hughes |
Los resultados se presentan como proporciones, porcentajes y medidas de tendencias centrales como media, mediana ± desviación estándar, y medidas de asociación como odds ratio con su respectivo intervalo de confianza del 95% (IC del 95%). La significación estadística entre las diferentes variables fueron analizadas mediante Fisher's exact test. La significación estadística fue establecida como p<0,05. El análisis estadístico se realizó mediante el software Epi Info 7.
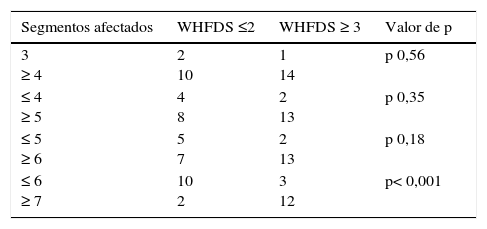
ResultadosEn nuestra muestra se identificó a 27 pacientes (N=27) con diagnóstico de LETM. El 74% (20/27) de sexo femenino. La edad media con sus respectivas desviaciones estándar fue de 38,22 ± 16 años. El 100% de los pacientes presentó trastornos sensitivos (superficiales y/o profundos) en al menos una extremidad y nivel sensitivo superficial se observó en el 92,59%. El sector más frecuente involucrado fue el dorsal (20/25) seguido del cervical (4/25) y, por último, el lumbar (1/25). Disfunción en el sistema motor fue observada en el 100%, siendo más frecuente en los miembros inferiores (52%). Los trastornos esfinterianos fundamentalmente vesicales (urgencia miccional, retención aguda de orina) se observaron en 25/27 pacientes. Respecto de las BOC, se realizaron en 19/27 pacientes y fueron positivas solo en 2 sujetos (10,52%). Los anticuerpos AQP4 fueron realizados en 18/27, siendo positivos en 6/18 (33,33%). Cuando analizamos las RM de encéfalo encontramos que 16/27 (59,25%) sujetos tenían RM normal. Dentro de las imágenes patológicas observamos que 6/27 (22,22%) tenían imágenes inespecíficas hiperintensas en T2 y FLAIR, el 14,81% (3/27) cumplía criterios para EM/ADEM y solo un paciente presentó lesiones compatibles con NMO (zonas con alta expresión de AQP4). Las lesiones en RM espinal fueron más frecuentemente observadas a nivel cervicodorsal (13/27) con una media de segmentos involucrados de 7,92 ± 4,2 y una mediana de 7,5. Cuando analizamos las causas encontramos que NMOSD representaba 10/27 (37,04%), de los cuales 8 tenían criterios de NMO definida y 2 presentaron LETM aislada con IgG-NMO positiva en suero (NMOSD limitada); idiopática 6/27 (22,22%); LES 3/27 (11,1%), de los cuales un paciente cumplía además criterio para síndrome antifosfolipídico (SAF); tumoral 3/27 (11,1%), de los cuales 2 fueron astrocitomas y uno MTS intramedulares; vascular 2/27 (7,4%), ambas fístulas durales; ADEM 2/27 (7,4%), y EM 1/27 (3,7%) (figura 1). Al 100% de los pacientes se les aplicó la WHFDS a los 3 meses y observamos que el 44,4% (12/27) requirió silla de ruedas y el 7,40% (2/27) al menos una ayuda para caminar. Un paciente presentó puntaje de 6 y falleció a los 3 meses por MTS intramedulares secundario a cáncer de pulmón. Cuando lo dividimos según necesidad de ayuda para caminar al menos 5 m (≥ 3), encontramos 15 sujetos, por consiguiente, ≤ 2 en 12 pacientes. Cuando comparamos cantidad de segmentos espinales vs. EFWH observamos que los pacientes con ≥ 7 segmentos presentaban peor pronóstico funcional (IC del 95%, odds ratio 20,0, p < 0,001) (véanse las tablas 2 y 3).

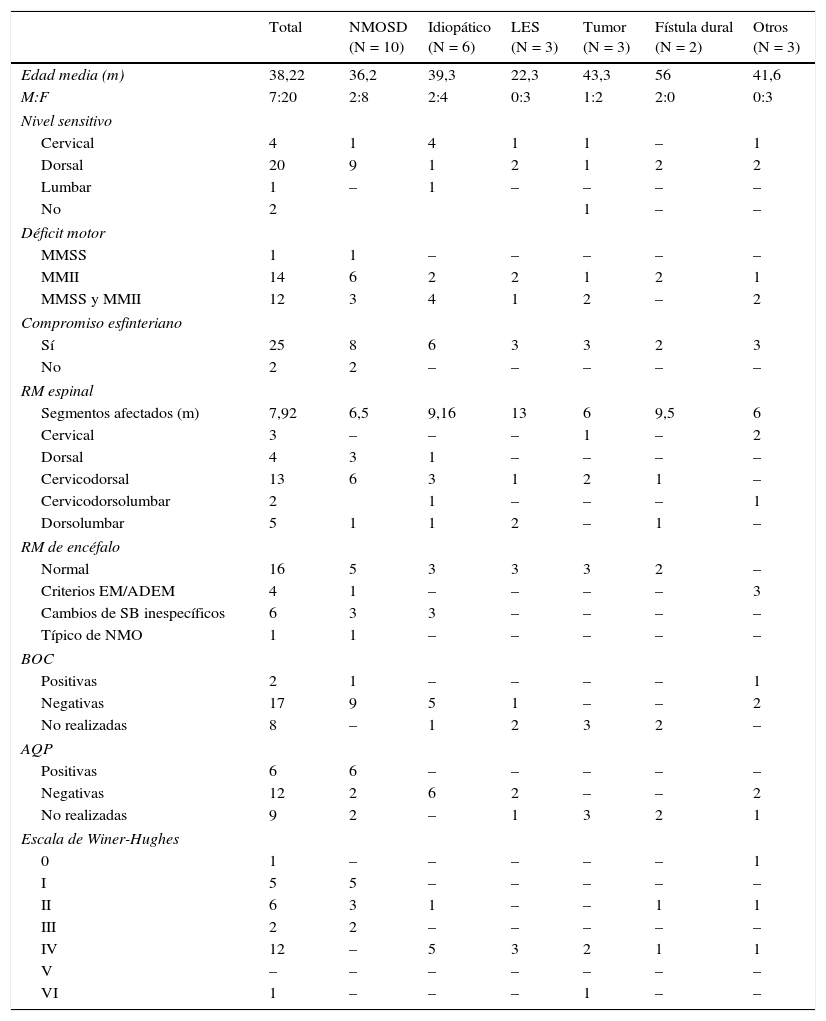
Características demográficas, clínicas, paraclínicas, neuroimágenes y pronóstico de la serie
| Total | NMOSD (N = 10) | Idiopático (N = 6) | LES (N = 3) | Tumor (N = 3) | Fístula dural (N = 2) | Otros (N = 3) | |
|---|---|---|---|---|---|---|---|
| Edad media (m) | 38,22 | 36,2 | 39,3 | 22,3 | 43,3 | 56 | 41,6 |
| M:F | 7:20 | 2:8 | 2:4 | 0:3 | 1:2 | 2:0 | 0:3 |
| Nivel sensitivo | |||||||
| Cervical | 4 | 1 | 4 | 1 | 1 | – | 1 |
| Dorsal | 20 | 9 | 1 | 2 | 1 | 2 | 2 |
| Lumbar | 1 | – | 1 | – | – | – | – |
| No | 2 | 1 | – | – | |||
| Déficit motor | |||||||
| MMSS | 1 | 1 | – | – | – | – | – |
| MMII | 14 | 6 | 2 | 2 | 1 | 2 | 1 |
| MMSS y MMII | 12 | 3 | 4 | 1 | 2 | – | 2 |
| Compromiso esfinteriano | |||||||
| Sí | 25 | 8 | 6 | 3 | 3 | 2 | 3 |
| No | 2 | 2 | – | – | – | – | – |
| RM espinal | |||||||
| Segmentos afectados (m) | 7,92 | 6,5 | 9,16 | 13 | 6 | 9,5 | 6 |
| Cervical | 3 | – | – | – | 1 | – | 2 |
| Dorsal | 4 | 3 | 1 | – | – | – | – |
| Cervicodorsal | 13 | 6 | 3 | 1 | 2 | 1 | – |
| Cervicodorsolumbar | 2 | 1 | – | – | – | 1 | |
| Dorsolumbar | 5 | 1 | 1 | 2 | – | 1 | – |
| RM de encéfalo | |||||||
| Normal | 16 | 5 | 3 | 3 | 3 | 2 | – |
| Criterios EM/ADEM | 4 | 1 | – | – | – | – | 3 |
| Cambios de SB inespecíficos | 6 | 3 | 3 | – | – | – | – |
| Típico de NMO | 1 | 1 | – | – | – | – | – |
| BOC | |||||||
| Positivas | 2 | 1 | – | – | – | – | 1 |
| Negativas | 17 | 9 | 5 | 1 | – | – | 2 |
| No realizadas | 8 | – | 1 | 2 | 3 | 2 | – |
| AQP | |||||||
| Positivas | 6 | 6 | – | – | – | – | – |
| Negativas | 12 | 2 | 6 | 2 | – | – | 2 |
| No realizadas | 9 | 2 | – | 1 | 3 | 2 | 1 |
| Escala de Winer-Hughes | |||||||
| 0 | 1 | – | – | – | – | – | 1 |
| I | 5 | 5 | – | – | – | – | – |
| II | 6 | 3 | 1 | – | – | 1 | 1 |
| III | 2 | 2 | – | – | – | – | – |
| IV | 12 | – | 5 | 3 | 2 | 1 | 1 |
| V | – | – | – | – | – | – | – |
| VI | 1 | – | – | – | 1 | – | – |
ADEM: encefalomielitis diseminada aguda; AQP: anticuerpos antiacuaporina 4; BOC: bandas oligoclonales; EM: esclerosis múltiple; F: femenino; m: media; M: masculino; MMII: miembros inferiores; MMSS: miembros superiores; NMO: neuromielitis óptica; RM: resonancia magnética.
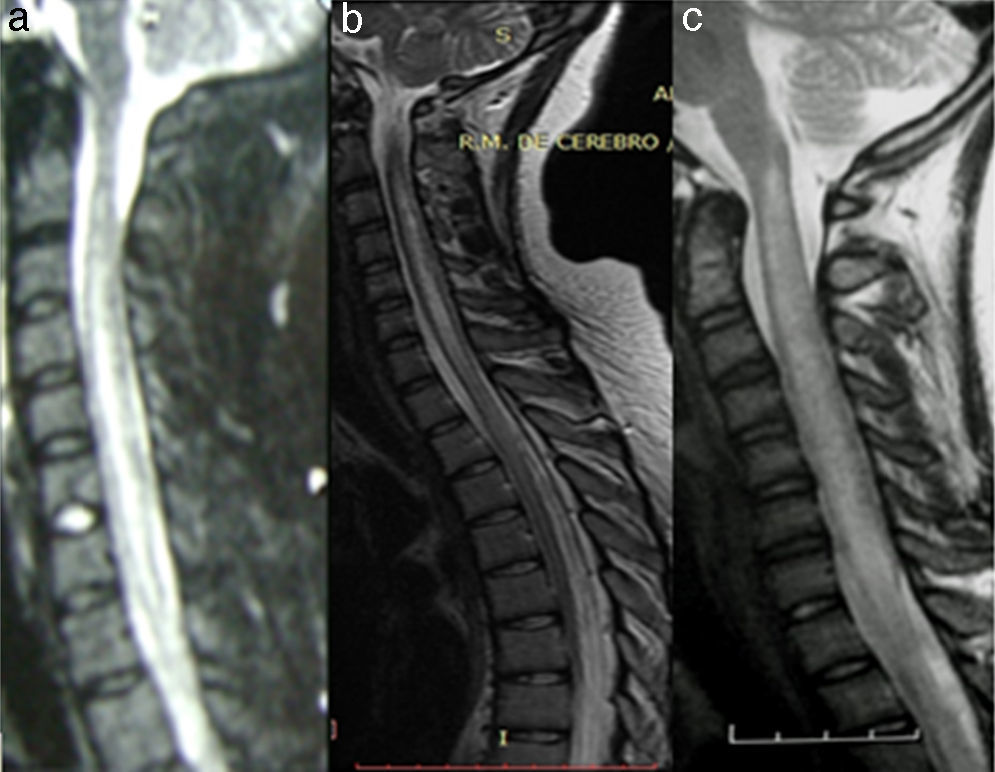
En nuestra serie, de una cohorte retrospectiva, encontramos a 27 pacientes con diagnóstico de LETM (véanse ejemplos en la figura 2). En concordancia con la literatura, el sexo más frecuente fue el femenino y la edad media fue de 38,22 años. Es interesante aclarar que la causa vascular fue de aproximadamente 10 años mayor (media: 56 años) que para el resto de las etiologías, como se observó en 2 estudios donde el sexo femenino y la edad menor de 40 años se asociaban a mielopatías inflamatorias en comparación con las causas vasculares16,17. La etiología más frecuente en nuestra serie fue la NMO (36%), seguida de la idiopática (22,2%) y solo encontramos un paciente con EM (3,7%) (véase resumen en la tabla 2). Un trabajo realizado en el Reino Unido18 informó que, de 76 pacientes con LETM (44 seropositivos para AQP4 y 32 seronegativos), 53 tenían diagnóstico de NMO (44 seropositivos para AQP4, 4 anti-MOG positivo y 5 «verdaderos» seronegativos), 5 ADEM, 5 idiopáticos, 4 infecciosas (uno tuberculosis, uno Mycoplasma, uno virus de Epstein-Barr, uno virus West Nile), 3 EM, 2 LETM asociado a anti-MOG positivo, 2 otras inflamatorias, una paraneoplásica y una vascular. Asimismo, en un estudio realizado en Brasil 19,NMO representó el 46,8%, LETM recurrente: 14%, LETM aislada: 16%, fístula AV: 4,4% y enfermedad reumática: 4,4% En dicho estudio, el 44% de las LETM recurrentes y el 20% de las aisladas presentaron seropositividad para AQP4. El resto de los diagnósticos etiológicos fueron seronegativos para IgG-NMO15. Estos hallazgos se correlacionan con los datos obtenidos en nuestro estudio19,20. Sin embargo, en un estudio retrospectivo multicéntrico21 (España y Francia) sobre 72 pacientes las causas de LETM fueron idiopática 22, EM 18, parainfecciosa 11, enfermedad sistémica 9, NMO 3, vascular 3, ADEM 2, fístula dural 2 y tumor 2. Además, un estudio realizado en la zona oeste de Australia22 mostró que LETM fue la presentación inicial en 13 de 26 pacientes evaluados (una monofásica, 4 EM, 3 NMO y 5 recurrente), mientras que en los restantes 13 pacientes (9 EM y 4 NMO) LETM ocurrió durante el seguimiento (1-27 años). Llama la atención la heterogeneidad etiológica según las diferentes poblaciones/regiones estudiadas. Ambos estudios muestran una mayor tasa de EM y menor frecuencia de NMO, lo que difiere de lo publicado en nuestra región.
 Imagen hiperintensa extensa en una paciente con ADEM. B) Imagen hiperintensa extensa en una paciente NMO seropositiva. C) Imagen hiperintensa extensa en una paciente con LES.")
Las manifestaciones clínicas son ampliamente conocidas y según diversas series no nos acercan al diagnóstico etiológico. Con respecto a los métodos complementarios de laboratorio, encontramos que en el LCR las BOC fueron realizadas en el 70,37% y fueron positivas en el 10,52% (2/19) de los pacientes realizados. Su positividad fue observada en una paciente con NMO y en el único sujeto con EM. Diversos estudios muestran su importancia en aquellos pacientes con primer episodio de mielitis atípica para EM, así como en el pronóstico y la conversión a EM clínicamente definida23,24. La positividad en la NMO está descrita entre el 15-30%4, similar a obtenido en nuestra población. En el año 2004, Lennon et al.25 describieron un anticuerpo llamado IgG-NMO (AQP4) sensible y específico para NMO y no para EM. En nuestra serie, los anticuerpos AQP4 se realizaron en el 66,66% y fueron positivos en el 75% de las NMO (en 2 pacientes no se realizó por falta de disponibilidad y dado que ya cumplían criterios para NMO definida). Estos anticuerpos tienen fundamental importancia en aquellos pacientes con primer episodio de LETM no solo para el diagnóstico de NMOSD (formas limitadas), sino para el pronóstico como muestra el trabajo inicial de Weinshenker et al., donde se observó que el 56% de las LETM seropositivas a los 2 años se convierten a NMO definida. Esto lleva a la necesidad del inicio precoz del tratamiento inmunosupresor para evitar nuevos episodios de mielopatía y/o neuritis óptica26-28. Destacamos la falta de disponibilidad para la detección de anticuerpos IgG- NMO y seguimiento a largo plazo, motivo por el cual fue difícil determinar la proporción real de pacientes con LETM que forman parte de NMOSD. Sin embargo, vale decir que a la totalidad de los pacientes con LETM idiopáticas se les efectuó dicho estudio, con resultados negativos28,29. Luego de la evaluación clínica inicial, el diagnóstico etiológico es dificultoso. Asimismo, un estudio francés30 observó que luego de la evaluación inicial 101/170 (59,4%) presentaron una causa no definida de mielitis y luego del seguimiento 55/101 fueron diagnosticados como mielitis inflamatoria o desmielinizante, y en solo 49/170 (28,8%) pacientes se llegó al diagnóstico etiológico definitivo de mielitis idiopática, EM o NMO. En concordancia con la literatura, nosotros observamos que el 59,2% de los pacientes tenían RM de encéfalo normal. Asimismo, destacamos que fue patológica en aquellos pacientes con ADEM y EM, y en NMO/idiopática se encontró un 30 y un 50% de lesiones inespecíficas, respectivamente. Las lesiones en zonas típicas de AQP se observan en el 7-9% de las NMO/NMOSD y en nuestra serie observamos en un 10% (1/10)31-34. Con respecto a la funcionalidad de los pacientes a los 3 meses, mediante la WHFDS encontramos que el 44,4% de nuestra serie requirió silla de ruedas. La etiología tumoral, la vascular, la enfermedad sistémica (lupus) e la idiopática tuvieron un alto porcentaje, con un puntaje de 4 en la WHFDS. Nosotros encontramos una correlación con ≥ 7 segmentos afectados y la necesidad de ayuda para caminar (p < 0,001) y no así con menor número de segmentos afectados. Sin embargo, en un estudio reciente de 23 pacientes se observó que el número de segmentos espinales afectados en LETM no se relaciona con peor pronóstico pero sí con recurrencia7. A pesar de las limitaciones de nuestro trabajo por su metodología retrospectiva, falta de obtención de anticuerpos IgG-NMO y BOC en la totalidad de la muestra, falta de seguimiento a largo plazo, y comparación de la WHFDS inicial y a los 3 meses, nuestro estudio provee una aproximación sobre los aspectos clínicos, paraclínicos, etiológicos y el perfil demográfico de las LETM en una población general en 2 centros públicos de la ciudad de Buenos Aires. Existen escasos estudios clínicos relacionados con las LETM en Argentina. Creemos necesario conocer el tipo de población que manejamos a diario para poder comparar nuestros resultados con los previamente publicados en otros países. Además, debido a las implicancias terapéuticas, consideramos que es de suma importancia realizar una exhaustiva evaluación para diferenciar las posibles etiologías asociadas a la LETM.
ConclusionesLas etiologías más frecuentes en nuestra serie fueron las NMOSD, seguida de las idiopáticas. Los anticuerpos IgG-NMO tienen un papel muy importante en el diagnóstico diferencial. Las características clínicas no fueron útiles en la orientación de un diagnóstico etiológico. El pronóstico funcional en pacientes con ≥ 7 segmentos afectados fue malo. Sin embargo, en la mitad de los casos de NMOSD fue bueno. Es esencial mantener un alto índice de sospecha de las diferentes etiologías posibles y su gravedad, principalmente en las LETM tumorales, vasculares, idiopáticas y por LES, que pueden representar un subconjunto crítico con distinta terapéutica y pronóstico.
FinanciaciónNo recibimos financiación para este trabajo.
Conflicto de interésLos autores declaran no tener ningún conflicto de interés.

