


El diagnóstico etiológico es un objetivo primordial para el manejo clínico de cada paciente. Algunos niños con cuadros clínicos complejos son objeto de una lista interminable de exámenes, lo que se ha denominado “odisea diagnóstica”, y que, sin embargo, muchas veces no deja resultados concluyentes. En los últimos años, estamos siendo testigos de una verdadera revolución de la medicina genómica con la incorporación al ámbito clínico de tecnologías que prometen aumentar la capacidad de hacer diagnóstico y disminuir los tiempos. Con énfasis en pediatría, se actualizan conceptos sobre las principales ventajas y limitaciones del diagnóstico genómico, en contraposición con las metodologías usuales.
Etiological diagnosis is essential in the clinical management of individual patients. Some children with complex medical conditions are subjected to numerous testing, known as “diagnostic odyssey”, which often gives no conclusive results. In recent years, a revolution in genomic medicine is underway with the use of technologies that promise to increase the ability to make a diagnosis and reduce the time involved. The main advantages and limitations of genomic diagnosis, as opposed to usual methodologies are reviewed with an emphasis on Pediatrics.
Las enfermedades humanas tienen un componente genético reconocido, ya sea como factor causal (por ejemplo, trastornos monogénicos, de genes contiguos y cromosómicos) o como factor predisponente (por ejemplo, trastornos multifactoriales o complejos).
En el transcurso de la última década, con el impulso del Proyecto Genoma Humano (HGP, por sus siglas en inglés) hemos entrado en una revolución del diagnóstico molecular, donde han surgido avances tecnológicos que permiten analizar todo el genoma de una forma más rápida, de manera simultánea y con gran capacidad de resolución.
Es así como progresivamente, durante los últimos años, las técnicas genómicas comienzan a estar disponibles para uso clínico, y existe una amplia gama de ensayos moleculares específi que ofrecen la confi ción etiológica para alrededor de 4000 condiciones monogénicas durante el año 20141. En este contexto, los ensayos basados en análisis genómicos ya están impactando en diferentes campos de la medicina, como la salud reproductiva, la oncología, el trasplante de órganos, la inmunología y las enfermedades infecciosas2–7.
El presente artículo pretende resumir las potencialidades del diagnóstico molecular desde el punto de vista genómico para trastornos genéticos constitucionales y sus posibles implicancias en la práctica clínica pediátrica.
La importancia del diagnóstico etiológicoEn diversas circunstancias, muchos pacientes entran en una espiral de estudios en búsqueda de una etiología, lo que se ha llamado la “odisea diagnóstica”8. Un gran número de esos estudios pueden evitarse con un diagnóstico etiológico certero y oportuno. Sin embargo, esto último no siempre es factible, y se genera un retraso en el diagnóstico, lo que conlleva diversas consecuencias que exceden el ámbito clínico, afectando a las familias, y a la larga, sobrecargando el sistema de salud9–11.
Aunque los trastornos genéticos, individualmente son considerados infrecuentes en la población general, es reconocido que una significativa parte de los pacientes hospitalizados en unidades pediátricas complejas tienen enfermedades con un fuerte componente genético, con estadías recurrentes, más largas y con mayores gastos que los pacientes sin una condición preexistente12–14. En nuestro medio, las herramientas genéticas disponibles para estudios de estos pacientes son limitadas y no siempre son las más adecuadas para todos los casos. La experiencia reportada en otros países ha mostrado casos en que el aporte de la genómica ha permitido un diagnóstico etiológico insospechado, con potenciales implicancias en el manejo terapéutico15–18.
Estos datos no implican que en todos los casos se deba partir haciendo un estudio del genoma completo, pues nada puede reemplazar el juicio clínico basado en una anamnesis completa, que incluya una historia familiar adecuada, junto con un examen físico minucioso. Es así, como en el caso de un diagnóstico genético probable, uno debiera hacer el estudio mejor indicado para su confirmación, ya sea del ámbito citogenético (cariograma, hibridación fluorescente in situ o FISH específico) o molecular (secuenciación capilar Sanger del gen de interés, Multiplex Ligation-dependent Probe Amplification o MLPA específico). Sin embargo, si no se llega a corroborar un diagnóstico o bien en el caso de que el paciente tenga una condición no clasificable o con manifestaciones poco comunes, uno debiera plantearse la posibilidad de un estudio amplio del genoma y sin sesgos19.
Diagnóstico genético convencional y diagnóstico genómicoBuscando anomalías en la estructura del genomaDiagnóstico convencional: cariotipo, FISH y MLPA
El estudio genético rutinario y básico desde la década de 1970ha sido el cariograma con bandeo GTG, el cual permite confi mar o descartar principalmente anomalías cromosómicas numéricas, y en menor medida, anomalías cromosómicas estructurales, que deben involucrar desde 5 a 10 millones de pares de bases (5-10 megabases; 5-10 Mb) para poder ser detectables con resolución estándar de 400-550 bandas citogenéticas20,21. En la década de 1990 emergieron técnicas citogenéticas moleculares que permitieron detectar microdeleciones o microduplicaciones cromosómicas, es decir, pérdidas o ganancias de material genético bajo el umbral de visibilidad en el microscopio óptico (tabla 1)21,22. Aquí destaca la técnica de hibridación in situ fl orescente (FISH), que está disponible para un número limitado de patologías20. Adicionalmente, se han incorporado al arsenal de herramientas diagnósticas algunas técnicas de biología molecular más finas en búsqueda de microdeleciones o microduplicaciones, entre las cuales destaca la de MLPA (Multiplex Ligation-dependent Probe Amplification)23. A diferencia del cariotipo convencional, que es una mirada global a la información genética (genoma), tanto la técnica de FISH como la de MLPA y otras relacionadas están diseñadas para buscar de forma dirigida anomalías conocidas en ciertas regiones cromosómicas definidas; por lo tanto, se debe contar con una hipótesis diagnóstica.
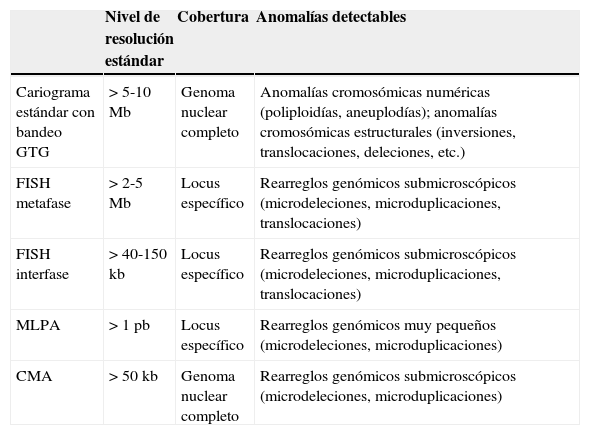
Comparación de los diferentes métodos de diagnóstico citogenético
| Nivel de resolución estándar | Cobertura | Anomalías detectables | |
|---|---|---|---|
| Cariograma estándar con bandeo GTG | > 5-10 Mb | Genoma nuclear completo | Anomalías cromosómicas numéricas (poliploidías, aneuplodías); anomalías cromosómicas estructurales (inversiones, translocaciones, deleciones, etc.) |
| FISH metafase | > 2-5 Mb | Locus específico | Rearreglos genómicos submicroscópicos (microdeleciones, microduplicaciones, translocaciones) |
| FISH interfase | > 40-150 kb | Locus específico | Rearreglos genómicos submicroscópicos (microdeleciones, microduplicaciones, translocaciones) |
| MLPA | > 1 pb | Locus específico | Rearreglos genómicos muy pequeños (microdeleciones, microduplicaciones) |
| CMA | > 50 kb | Genoma nuclear completo | Rearreglos genómicos submicroscópicos (microdeleciones, microduplicaciones) |
CMA: cariotipo molecular; FISH: hibridación in situ fluorescente; GTG: bandeo G por tripsina-giemsa; MLPA: Multiplex Ligation-dependent Probe Amplification.
Unidades: kb: kilobases; Mb: megabases; pb: pares de bases.
En la últimas décadas, los grandes avances en biotecnología cimentaron las bases para el desarrollo de nuevos métodos que permitieron optimizar el limitado nivel de detección de las técnicas existentes. Por consiguiente, se pudo mejorar la baja capacidad diagnóstica de las técnicas citogenéticas convencionales y moleculares disponibles.
Es así como la técnica de la hibridación genómica comparativa (CGH, por sus siglas en inglés)24, desarrollada en la década de 1990, recobró relevancia de la mano del desarrollo de biochips, llevando al ámbito diagnóstico los microarreglos (microarrays). Los conceptos de microarreglos cromosómicos o cariotipo molecular (CMA) implican diferentes plataformas tecnológicas que permiten analizar o interrogar el genoma completo de un individuo en un chip25. El uso de esta herramienta en el ámbito clínico permite la identifi ción de desequilibrios genómicos submicroscópicos con una resolución de hasta 50kb, lo que equivale a cientos de veces mayor resolución que un cariotipo con bandeo GTG convencional26,27. Los desequilibrios de tamaño submicroscópico y que resultan en pérdidas o ganancias de información genética sobre 1kb son denominados variaciones en el número de copias (CNV, por sus siglas en inglés)28,29. Los CNV son parte de la variabilidad interindividual, son relativamente comunes en la estructura de nuestros genomas y no necesariamente se asocian a una patología30.
Ventajas del diagnóstico con CMASi bien existen diferentes metodologías que permiten realizar un CMA, que se diferencian en la plataforma tecnológica utilizada y en el nivel de resolución alcanzado, la técnica ha permitido reconocer enfermedades genéticas causadas por CNV que no son fácilmente detectadas con los métodos anteriormente descritos, lo que aumenta la capacidad diagnóstica y la descripción de nuevos síndromes (tabla 2)31,32.
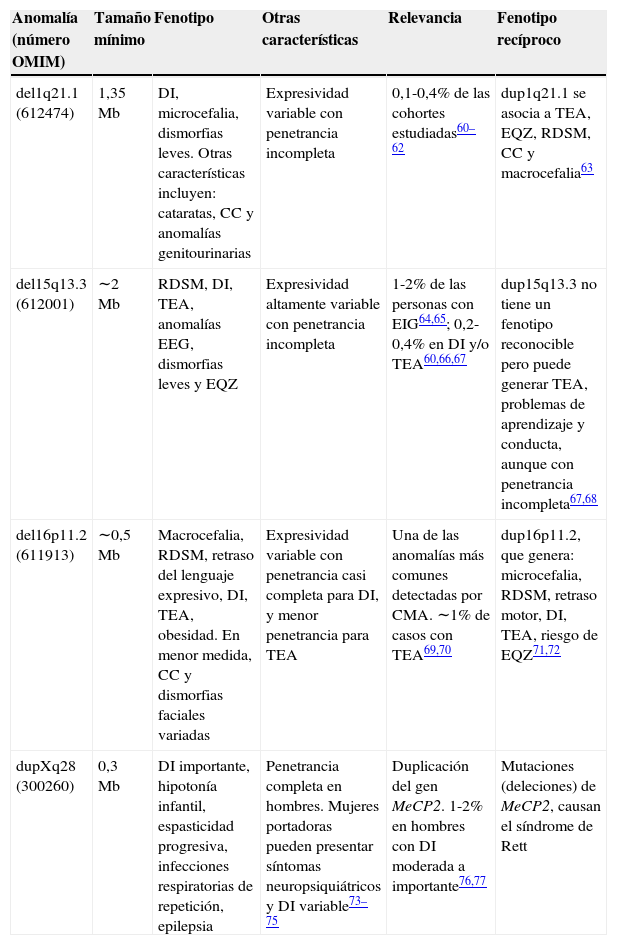
Ejemplos seleccionados de nuevos síndromes de microdeleción y microduplicación reconocidos a partir del uso del CMA en el ámbito clínico
| Anomalía (número OMIM) | Tamaño mínimo | Fenotipo | Otras características | Relevancia | Fenotipo recíproco |
|---|---|---|---|---|---|
| del1q21.1 (612474) | 1,35 Mb | DI, microcefalia, dismorfias leves. Otras características incluyen: cataratas, CC y anomalías genitourinarias | Expresividad variable con penetrancia incompleta | 0,1-0,4% de las cohortes estudiadas60–62 | dup1q21.1 se asocia a TEA, EQZ, RDSM, CC y macrocefalia63 |
| del15q13.3 (612001) | ∼2 Mb | RDSM, DI, TEA, anomalías EEG, dismorfias leves y EQZ | Expresividad altamente variable con penetrancia incompleta | 1-2% de las personas con EIG64,65; 0,2-0,4% en DI y/o TEA60,66,67 | dup15q13.3 no tiene un fenotipo reconocible pero puede generar TEA, problemas de aprendizaje y conducta, aunque con penetrancia incompleta67,68 |
| del16p11.2 (611913) | ∼0,5 Mb | Macrocefalia, RDSM, retraso del lenguaje expresivo, DI, TEA, obesidad. En menor medida, CC y dismorfias faciales variadas | Expresividad variable con penetrancia casi completa para DI, y menor penetrancia para TEA | Una de las anomalías más comunes detectadas por CMA. ∼1% de casos con TEA69,70 | dup16p11.2, que genera: microcefalia, RDSM, retraso motor, DI, TEA, riesgo de EQZ71,72 |
| dupXq28 (300260) | 0,3 Mb | DI importante, hipotonía infantil, espasticidad progresiva, infecciones respiratorias de repetición, epilepsia | Penetrancia completa en hombres. Mujeres portadoras pueden presentar síntomas neuropsiquiátricos y DI variable73–75 | Duplicación del gen MeCP2. 1-2% en hombres con DI moderada a importante76,77 | Mutaciones (deleciones) de MeCP2, causan el síndrome de Rett |
CMA: Cariotipo molecular; CC: cardiopatía congénita; DI: discapacidad intelectual; EIG: epilepsia idiopática generalizada; EQZ: esquizofrenia; MODY: Madurity-Onset Diabetes of the Young; OMIM: Online Mendelian Inheritance in Man; RDSM: retraso del desarrollo psicomotor; TEA: trastorno del espectro autista.
En este contexto, las últimas recomendaciones de diferentes sociedades científi la consideran la técnica de primera línea en el estudio de los trastornos del desarrollo, como anomalías congénitas múltiples no identifi como un síndrome reconocido, discapacidad intelectual aparentemente no sindrómica y en trastornos del espectro autista33–35.
Adicionalmente, existen microarreglos que incorporan la identificación de polimorfismos de nucleótido único (SNP, por las siglas en inglés), lo que permite identificar grandes regiones de homocigocidad, que pueden indicar que existe consanguinidad o casos de disomía uniparental, con implicancias en casos de posibles enfermedades recesivas.
Limitaciones del diagnóstico con CMAA pesar de ser una técnica poderosa, el enfoque con CMA solo permite detectar desequilibrios en la información genómica, por lo que aquellas anomalías que involucren un reordenamiento, pero no una pérdida o ganancia neta, pasarán desapercibidas para el método (por ejemplo, inversiones, translocaciones balanceadas). Por otro lado, las herramientas de uso clínico están enfocadas al conocimiento de la información genómica relacionada con problemas médicos, por lo que aquellas variaciones de zonas de heterocromatina de ciertos cromosomas que son visibles al cariotipo no serán detectadas con CMA (por ejemplo, tallos y satélites de cromosomas acrocéntricos que pueden configurar pequeños cromosomas marcadores supernumerarios). Adicionalmente, la mayoría de los algoritmos bioinformáticos tienen limitaciones para detectar poliploidías y bajos niveles de mosaicismo.
Otro aspecto importante a considerar es que en el análisis por CMA no se visualizan directamente los cromosomas y su estructura, por lo que esta técnica no es capaz, por sí sola, de dar información referente a la ubicación física de los desequilibrios, así como tampoco puede señalar un mecanismo patogénico y riesgo de recurrencia.
Otra limitación está relacionada con la interpretación de los CNV no reportados previamente. Sin embargo, el desarrollo de bases de datos de acceso público con CNV encontrados en individuos controles y en pacientes han ayudado a disminuir progresivamente los casos de incertidumbre36–38.
Por estos antecedentes, el cariotipo convencional debe seguir siendo la elección si se sospecha un trastorno como poliploidía o una aneuploidía (trisomías o monosomía), o si existe una historia familiar sugerente de que haya portadores de translocaciones balanceadas (abortos recurrentes, infertilidad, etc.).
De la misma forma, si se sospecha un síndrome de microdeleción o de genes contiguos con un test clínico disponible, sigue recomendándose realizar primero el FISH o MLPA específico.
Buscando anomalías en la secuencia del genomaDiagnóstico convencional: secuenciación de Sanger
Hasta hace unos años, la búsqueda de mutaciones puntuales para el diagnóstico específico de una determinada enfermedad monogénica estaba restringida principalmente al análisis de un locus o gen de interés por vez, bajo secuenciación capilar basada en el principio de Sanger39. Sin embargo, debido a que un número importante de enfermedades monogénicas presentan heterogeneidad genética, es posible que el análisis de un único gen sea insuficiente para detectar todas las posibles causas de una misma enfermedad y se requiera analizar varios genes de forma sucesiva y racionalizada hasta encontrar una etiología. Abordar este problema mediante secuenciación de Sanger incrementa los costos en términos económicos y de tiempo, lo que la hace ineficiente40.
Adicionalmente, la capacidad de hacer un diagnóstico etiológico en una condición monogénica estaba muy limitada en casos en que no existieran nuevas hipótesis diagnósticas o en presencia de fenotipos sin etiología reconocida.
Diagnóstico genómico: secuenciación de nueva generación
En los últimos 10 años, e impulsada por la fi alización del HGP, surgió una segunda generación de metodologías para permitir una secuenciación a gran escala llamada secuenciación de siguiente o nueva generación (NGS, por sus siglas en inglés)41. Existen diferentes plataformas basadas en NGS, que se han ido incorporando al ámbito clínico, aumentando el poder de diagnóstico, principalmente debido a la posibilidad de hacer una gran cantidad de análisis de forma paralela, con menores costos y mayor rendimiento que su predecesora, la secuenciación de Sanger.
Tan solo un par de años después de haber finalizado el HGP, ya estaba en el mercado el primer equipo de NGS42. Actualmente, para uso clínico está disponible la secuenciación por NGS de genes individuales, de paneles o conjuntos de genes, así como el conjunto de todas las secuencias de ADN que codifican proteínas (exones), conocido como exoma. Debido a que gran parte de nuestro genoma no codifica para alguna proteína, la secuenciación del exoma completo (WES) abarca aproximadamente el 1-2% del total del genoma humano. Como es sabido que no todas las variantes patogénicas se encuentran en las secuencias codificantes de proteínas, sino que también aparecen en secuencias que tienen una función regulatoria de la expresión génica, se está avanzado cada vez más hacia el uso clínico de la secuenciación del genoma completo (WGS), que ofrece una cobertura de entre un 85 y un 95% del total de la secuencia de un genoma humano19.
Como se comentó, actualmente existen variadas plataformas de NGS, aunque el equipo MiSeqDx de Illumina es el único sistema de diagnóstico in vitro aprobado por la Food and Drug Administration (FDA) de los EE. UU43. Este equipo es un secuenciador de NGS de sobremesa diseñado específicamente para pruebas de diagnóstico. De hecho, el MiSeqDx se utiliza actualmente para el diagnóstico molecular de la fibrosis quística. Para esto dispone de dos kits; si bien ambos implican secuenciar el gen CFTR, difi ren en lo que están secuenciando. En el primer kit se secuencian 139 variantes comunes en la población caucásica y en el segundo se secuencia por completo el gen CFTR. De esta manera, este ensayo clínico para el gen CFTR es el primer sistema de diagnóstico basado en NGS en el mercado aprobado por la FDA44.
Ventajas del diagnóstico mediante NGSLas ventajas del uso de NGS para diagnosticar enfermedades, y especialmente trastornos genéticos, son múltiples45. Un test de NGS proporciona la misma o más información que la realización de múltiples test basados en secuenciación por Sanger, pero es más rápido y tiene un precio significativamente más bajo por base secuenciada46. Esto se debe a que la tecnología de NGS permite “interrogar” a muchos nucleótidos (por ejemplo, paneles, WES) o todos los nucleótidos del genoma (por ejemplo, WGS) simultáneamente. Además, el uso de los llamados barcode o códigos de barra (secuencias específicas de nucleótidos incorporados en los extremos de los fragmentos a secuenciar) permite procesar múltiples muestras de muchos pacientes a la vez41. Otra de las ventajas es que los genes que se consideraban raramente implicados en una enfermedad y que por lo tanto no se justificaba el costo de desarrollo de un test clínico, pueden ahora ser fácilmente incorporados en paneles de genes para enfermedades específicas sin aumentar considerablemente el costo de la prueba46.
Por otra parte, la secuenciación masiva permite interrogar al genoma y obtener múltiples tipos de información útil para un diagnóstico. En este contexto, puede detectar SNP, inserciones/deleciones (indels), CNV y translocaciones. Utilizando la misma técnica, pero empleando ARN como material de partida, se puede evaluar transcripción de ARNm y/o micro-ARN, entre otros41.
Limitaciones del diagnóstico NGSPor supuesto, como con cualquier tecnología, también hay limitaciones para el diagnóstico de NGS.
Desde el punto de vista técnico, aunque la tecnología se destaca en la identificación de mutaciones raras, es menos eficiente en la búsqueda de indels que tienen más de 8-10 pares de bases de longitud19. Asimismo, no es capaz por sí sola de identificar mutaciones inestables por expansión de nucleótidos, ni alteraciones epigenéticas19, las cuales deben ser corroboradas por otra técnica independiente. Por otra parte, dependiendo de la cobertura y profundidad del análisis, puede haber bajo nivel de lecturas sobre algunas regiones del genoma, lo que puede significar una pérdida de información putativamente relevante40.
También hay desafíos desde el punto de vista bioinformático en el análisis de los resultados producidos por NGS, más específicamente en términos de almacenamiento, manipulación e interpretación de datos3. Los principales inconvenientes de NGS están relacionados con la inmensa cantidad de datos que se generan, lo que provoca verdaderos cuellos de botella en el análisis y la interpretación de un gran número de variantes no reportadas previamente, con el consiguiente impacto en los costos y en el tiempo de respuesta. El desconocimiento de la magnitud de la variación genética normal puede llevar a la confusión en cuanto al verdadero significado de las nuevas variantes, información que puede confundir a los tratantes y los pacientes. Estos inconvenientes pueden resolverse mediante la realización simultánea de este tipo de estudio en los padres del probando y en otros familiares afectados47, así como el aumento de la comunicación y el intercambio de datos entre los laboratorios a través de bases de datos públicas, como ClinVar48.
Se debe considerar que WES tiene una tasa de detección de un 25% para variantes patogénicas49, por lo que debe procurarse una juiciosa selección de los pacientes y racionalmente agotar las instancias previas según cada caso. Al igual que en el caso de CMA, si uno tiene algún gen candidato, es mejor partir haciendo el análisis estándar. Si se trata de un fenotipo con varios genes posibles, sería más útil considerar la realización por NGS de un panel de los genes conocidos. Si no se encuentra mutación en los genes probables o no hay una orientación clínica del posible gen involucrado, tendría más utilidad considerar un WES o WGS. Por costos, WES puede ser más costo-efectiva que WGS, pero hay que considerar que un resultado negativo no descarta que pueda haber una mutación en una secuencia no codificante.
Es importante indicar que los costos han bajado notablemente desde las primeras experiencias de secuenciación completa del genoma humano, y se espera que sigan disminuyendo. Sin embargo, en el caso de WGS, el costo ronda los 10 000 dólares50. Los esfuerzos están enfocados en obtener ensayos que bordeen los 1000 dólares, aunque por el momento es una meta que algunos investigadores creen difícil de conseguir por los cuellos de botellas de la interpretación y el análisis de los datos51.
Nuevos desarrollos clínicos basados en el diagnóstico genómicoA pesar de las dificultades, los avances en las pruebas genómicas ya están impactando directamente en la vida de las personas. El ejemplo más notable de la potencia de NGS para revolucionar todo un campo de la medicina ha sido la rápida adopción, desde finales de 2011, de la prueba prenatal no invasiva para la detección de aneuploidías fetales en embarazos de alto riesgo2.
Otros avances que pueden afectar a la evolución de casos de difícil manejo clínico son las diversas experiencias reportadas por centros de los EE. UU., que ofrecen diagnóstico de NGS en plazos breves18. Un ejemplo de ello es la prueba genética llamada STAT-Seq, del Centro de Medicina Genómica Pediátrica implementada en la unidad de cuidados intensivos neonatal en el Children's Mercy Clinical Hospital de Kansas City52,53. El principio de STAT-Seq se basa en reducir el tiempo de diagnóstico a alrededor de dos días utilizando WGS con un flujo de análisis (pipeline) bioinformático automatizado. Habitualmente, las familias de los recién nacidos con enfermedades genéticas esperan semanas, meses o incluso años para tener la oportunidad de recibir un diagnóstico. Dada la existencia de más de 4000 enfermedades genéticas diferentes, muchas familias se quedan sin diagnóstico etiológico, incluso con la realización de diferentes test específicos. En otros casos, hay familias que reciben un diagnóstico post mórtem2. Con STAT-seq y otras experiencias similares, se pueden tomar decisiones informadas sobre las mejores alternativas de manejo y seguimiento disponibles con el conocimiento actual, e incluso existe la posibilidad de identificar una enfermedad potencialmente tratable18,52,53.
Estos son solo algunos ejemplos, ya que diversos centros y compañías han desarrollado test con paneles de genes (es decir, se selecciona un grupo de genes que se sabe están relacionados de alguna manera con la enfermedad) en patologías tan diversas como: cáncer, enfermedades neurológicas, enfermedades cardíacas, enfermedades infecciosas y dermatológicas, entre otras54–58. Hablar de cada una de ellas se escapa del interés de este artículo; sin embargo, resulta interesante ver cómo cada día se van sumando nuevas patologías al diagnóstico de NGS.
Si bien el diagnóstico molecular basado en NGS está empezando a tomar fuerza, aún queda mucho por hacer e implementar, tanto a nivel de estrategias de secuenciación como a nivel de interpretación y análisis de los resultados, tanto a nivel de cómo filtrar e identificar las variantes (análisis bioinformático) así como en la interpretación clínica. En este contexto, se han desarrollado diversos algoritmos informáticos que permiten detectar grandes variaciones estructurales con los datos de NGS. Dependiendo del perfeccionamiento de estas herramientas se podría lograr a medio plazo que un único ensayo tenga la capacidad de detectar de forma simultánea rearreglos genómicos de gran escala y cambios puntuales29.
Pese a las dificultades, en los años venideros contaremos con una creciente cantidad de test que utilizarán este tipo de tecnología.
ConclusionesEs innegable que cada vez se ha avanzado más en el reconocimiento de las bases moleculares de las enfermedades humanas. Actualmente, estos avances están disponibles para el uso clínico, y entre ellos destaca la existencia de análisis moleculares que permiten la confirmación de las mutaciones.
Un punto no menos importante es el costo de estas herramientas diagnósticas. En varios países, el CMA está incorporado en las coberturas de los sistemas de salud y puede tener un costo similar al de un cariotipo convencional. En el caso de NGS, su costo ha bajado notablemente desde la finalización del HGP, aunque aún sigue siendo prohibitivo para gran parte de la población. En nuestro país, solo un puñado de estudios genéticos tiene algún tipo de cobertura dentro del sistema de salud, por lo que existe el desafío de que haya una actualización en la lista de estudios y que vaya a la par con los nuevos avances y las crecientes necesidades.
Debido a que es esperada una variabilidad genética individual, existen muchos desafíos relacionados con la interpretación de los hallazgos. En el caso del CMA hay cada vez más experiencia acumulada, con disponibilidad de bases de datos de variaciones normales y patogénicas, lo que ayuda a que cada vez existan menos casos de incierta interpretación. En el caso de la NGS, junto con bases de datos específicas, muchas veces es necesario hacer ensayos complementarios (funcionales o modelamientos informáticos) para disponer de información clara sobre la posible patogenicidad de una variante.
Por lo anterior, es importante reafi que, como para cualquier test genético, los pacientes y sus familias deben tener un asesoramiento antes y después del test donde se pueda conversar sobre los posibles escenarios que puede generar esta información, lo que se conoce como asesoramiento genético. El conocimiento derivado de identifi la etiología permite acortar la “odisea diagnóstica”, asesorar adecuadamente a los pacientes y sus familias en la toma de decisiones, así como optimizar los escasos recursos existentes, centrándose en los problemas conocidos y previsibles según cada patología. En algunos casos, el conocimiento del tipo de mutación puede tener implicancias no solo en el diagnóstico, sino también en el manejo, seguimiento y en potenciales perspectivas terapéuticas59.
Glosario de términos de genética utilizado en el escritoCariotipo molecular (CMA) omicroarrayscromosómicos. Técnica basada en microchips que permite detectar pérdidas o ganancias de información genómica de una muestra (linfocitos, tumor, etc.) de un individuo en comparación con un control o referencia.
Copy-number variants(CNV). Es un tipo de variación genética submicroscópica que implica pérdida o ganancia de información a partir de 1kb. Si un CNV se encuentra sobre el 1% de la población, es considerado polimórfico (copy-number polymorphism,CNP).
Exoma. Conjunto total de exones. Por tanto, es el conjunto de información genética que codifica para proteínas, lo que corresponde a aproximadamente el 1% del genoma nuclear humano.
Genoma. La totalidad de la información genética contenida en una célula de un organismo determinado. En la especie humana debemos considerar el genoma nuclear (conjunto de los cromosomas) y el genoma extranuclear (ADN mitocondrial).
Indel. Inserción o deleción de uno o más nucléotidos. Cualquier indel con un tamaño sobre 1kb se debe considerar CNV.
Secuenciación de Sanger. Método ideado por F. Sanger para obtener la secuencia ordenada de nucleótidos de fragmentos relativamente pequeños de ADN (∼500 pb). Sigue siendo el método de referencia (gold standard) para corroborar la existencia de mutaciones puntuales detectadas por NGS.
Next-generation sequencing(NGS). Diversas metodologías que permiten obtener secuencias genómicas a gran escala (desde locus/genes individuales hasta genomas completos), en un menor plazo y con menor costo total que si se realizara con la secuenciación de Sanger.
Heterogeneidad genética (delocus). Fenómeno en que un mismo fenotipo puede ser la consecuencia de anomalías en locus (genes) diferentes. Es decir, en un individuo o familia, la enfermedad “A” es causada por el gen “Z”, mientras que en otra familia la misma enfermedad “A” es causada por el gen “W”. Muchas enfermedades genéticas presentan este fenómeno, incluso hay enfermedades que pueden ser generadas por mutaciones en más de 10 genes distintos y con patrones de herencia diferentes.
Single nucleotide polymorphism(SNP).Locus de un único nucleótido, donde es habitual que existan dos alelos posibles (ejemplo: o nucleótido A o bien nucleótido G). Si el alelo (nucleótido) menos común se encuentra sobre el 1% de una población se considera SNP, mientras que si el alelo minoritario se encuentra en menos del 1%, este alelo es una variante rara (single nucleotide variant, SNV). Se conocen varios millones de SNP a lo largo de todo el genoma humano, por lo que buena parte de la variabilidad normal entre individuos viene dada por este tipo de polimorfismo.
Whole exome sequencing(WES). Metodología que permite estudiar la secuencia completa del exoma.
Whole genome sequencing(WGS). Metodología que permite estudiar la secuencia completa del genoma. Incluye al exoma, así como a las secuencias intrónicas e intergénicas.
Conflicto de interésEste trabajo cumple con los requisitos sobre consentimiento/asentimiento informado, comité de ética, financiamiento, estudios animales y sobre la ausencia de confl os de intereses según corresponda.
