



Neutrophils play an important role in immune defence against several pathogens. These cells actively participate in the innate immune response through different functions, such as chemotaxis, phagocytosis, oxidative burst and degranulation, which have been widely studied. However, in the last few years, a new function has been described; activated neutrophils are able to release web-like chromatin structures known as neutrophil extracellular traps (NETs). These structures formed by DNA, histones, and proteins, immobilize and kill microorganisms. Disruption in NET formation is associated with the pathophysiology of several disorders, including the autoimmune diseases. NETs are an important source of the autoantigens involved in the production of autoantibodies and maintenance of the inflammatory milieu. This review provides a summary of the contribution of NETs to the pathogenesis of anti-neutrophil cytoplasmic antibodies-associated vasculitis, systemic lupus erythematosus, and rheumatoid arthritis. The preliminary findings on NETs components in Sjögren's syndrome will also be described.
Los neutrófilos juegan un papel muy importante en la defensa inmune contra diferentes patógenos. Estas células participan activamente en la respuesta inmune innata a través de diferentes funciones como quimiotaxis, fagocitosis, estallido oxidativo y degranulación, las cuales han sido estudiadas ampliamente. Sin embargo, en los últimos años se ha descrito una nueva función; los neutrófilos activados son capaces de liberar redes de cromatina llamadas trampas extracelulares de neutrófilos (NETs). Estas estructuras están formadas por ADN, histonas y proteínas capaces de inmovilizar y matar microorganismos. Alteraciones en la formación de estas NETs están asociadas con la fisiopatología de varios trastornos, incluyendo las enfermedades autoinmunes (EAI). Las NETs son consideradas una fuente de autoantígenos que ayudan a la producción de autoanticuerpos y al mantenimiento de un ambiente inflamatorio. Esta revisión resume la contribución de las NETs a la patogénesis de vasculitis asociada a anticuerpos contra el citoplasma de los neutrófilos, lupus eritematoso sistémico y artritis reumatoide. Adicionalmente, se describirán los resultados preliminares de la detección de componentes de las NETs en pacientes con síndrome de Sjögren.
Neutrophils are the most abundant immune cells involved in the antimicrobial innate response, which is articulated through three main mechanisms: phagocytosis, degranulation and the formation of neutrophil extracellular traps (NETs). NET formation is the consequence of a process named NETosis, which occurs when activated neutrophils release extracellular structures made up of decondensed fibers of chromatin and cytoplasmic, granular, and nuclear elements, which can trap and kill bacteria, fungi, viruses, and parasites.1–3 Although NET formation could be considered a benefit to the host, evidence demonstrates a strong association with NET production and several pathologies such as sepsis, atherosclerosis, cancer, and autoimmunity.4
Regarding autoimmunity, mechanisms involved in the breakdown of self-tolerance includes the development of pathogenic autoantibodies. The fact that many of the molecular elements exteriorized by the NETs include autoantigens recognized as foreign during the autoimmune process (i.e., myeloperoxidase (MPO), double-strand DNA (ds-DNA) and histones), suggests that the exposure of such autoantigens to immune cells plays a plausible role in the development of an abnormal immune response in susceptible individuals.5 Autoantibodies and inflammation prompted by NETs could lead to tissue damage and the release of new autoantigens, thus creating new targets for autoimmune responses and promoting the introduction of a vicious autoimmune cycle.5 Moreover, aberrant NET formation has been observed in patients with autoimmune disease (ADs), which argues for their implication in the autoimmune pathology.6,7
In this review, the role of NET formation in systemic ADs such as anti-neutrophil cytoplasmic antibodies (ANCA)-associated vasculitis (AAV), systemic lupus erythematosus (SLE), and rheumatoid arthritis (RA) will be assessed. In addition, preliminary findings on NETosis components in patients with Sjögren's syndrome (SS) will be described.
NETs in ANCA-associated vasculitis (AAV)AAV includes different autoimmune disorders (e.g., granulomatosis with polyangiitis, eosinophilic granulomatosis with polyangiitis and microscopic polyangiitis) characterized by an immune cell infiltration and necrosis of small or medium blood vessels. A hallmark of AAV is the presence of autoantibodies against neutrophil-associated proteins such as MPO, proteinase-3 (PR3) and/or the lysosomal membrane protein-2 (LAMP-2).8,9 An increasing amount of evidence links the NET formation with AAV pathology. Indeed, Kessenbrock et al.6 found depositions of NETs in kidney biopsies from AAV patients with high neutrophil infiltrations. Moreover, they showed the co-location of MPO and PR3 within the chromatin web of the NETs, thus suggesting the continuous autoantigen stimulation of immune cells involved in autoantibody production. In addition, they found in vitro that ANCA IgG-stimulated neutrophils were able to induce sustainably NET formation as compared to neutrophils stimulated with IgG antibodies from the control group. Along the same line, histopathologic staining of kidney biopsies from an AAV patient with glomerulonephritis showed NET presence in the necrosis site in both the glomerular capillary tuft and the interlobular artery, thus suggesting that NET formation has a detrimental effect on vasculitis.10
A recent study compares the frequency of NETs in AAV with non-ANCA vasculitis. They found a more abundant NET presence in AAV than in other vasculitides, and NETs from AAV patients were more resistant to DNase degradation.11 Interestingly, a study on 99 AAV patients showed that NET formation was not correlated with serological levels of ANCAs, but they were with activity of disease. Thus, NETs were prominently present in relapsing hospitalized patients as compared to AAV patients in remission.12 In order to clarify the mechanism by which NET formation can induce AAV development, Sangaletti et al.7 transferred dendritic cells (DCs) uploaded with NET components to naïve mice and showed the production of ANCA antibodies in the receptor mice. Interestingly, when NETs were pre-treated with DNase, the autoimmunity was not induced in the naïve group.
Mechanism by which NET formation induces tissue damage in AAV was elucidated by Schreiber et al.13 They demonstrated in vitro that NETs from ANCA-activated neutrophils induce permeabilization of the endothelial monolayer. This damage was reduced when neutrophils were treated with DNase-I or NET inhibitor. Furthermore, they found that NETosis activates the alternative pathway of the complement leading to the formation of the membrane attack complex. This discovery was tested in vivo in a murine model in which mice treated with anti-MPO IgG (capable of activating neutrophils and inducing NET formation) were evaluated. Seven days post-challenge, mice developed a glomerular endothelial injury showing histological necrosis which was blunted by DNase treatment, thus demonstrating the role of NET formation in tissue damage.13 Another study demonstrated that NETs are not exclusively involved in innate immune dysregulation but also induce adaptive immune dysregulation. Peripheral blood mononuclear cells from healthy donors were stimulated with NETs from AAV patients. Interestingly, B-cells and T-cells proliferate upon stimulation along with an increase in interleukin (IL)-17 secretion, thus contributing to AAV pathology.14
Another comorbidity associated with AAV is thrombotic events. NETs are also implicated in this phenomenon since they can cause thrombosis via platelet activation and aggregation.15 A case report from 2012 showed abundant NET formation within the thrombus from a fatal case of AAV as compared to thrombi from non AAV patients.16 Another study demonstrated that NETs by themselves without activation of platelets were able to induce thrombotic events. Indeed, neutrophils primed with the complement protein C5a formed NETs that bear tissue factor on the chromatin web. This factor was able to activate the coagulation system.17,18
Given all of the above-mentioned data, treatment aiming to block or degrade NETs is necessary to treat NET-induced complications in AAV patients.5 For instance, Rituximab, an FDA-approved treatment for AAV, has shown efficacy in AAV remission.19 It is likely that depletion of B-cells and reduction of ANCAs contribute to less antibody-mediated neutrophil activation and a decrease in NET formation. Other treatments such as MPO blockade,20 anti-C5 receptor,17,21 or DNase treatment in vitro and in vivo (in murine models) have shown their efficacy in reducing NETs11; nevertheless, no data on AAV patients is available. Along the same line, blockade of phosphoinositide 3-kinase gamma, a kinase implicated in the neutrophil activation pathway, in a murine model leads to AAV symptom alleviation.22 Clinical trials leading to NET suppression in AAV patients are warranted.
NETs in systemic lupus erythematosus (SLE)The pathogenesis of SLE involves the defective clearance of immune complexes and debris with nucleic acids, excessive activation of the innate immune system (i.e., toll-like receptor (TLR) and type I interferon (IFN-I)), and activation of aberrant lymphocytes.23 NETs activate the innate immune response of plasmacytoid DCs through TLR9. In addition, patients with SLE develop autoantibodies against DNA and NET antimicrobial peptides, thus demonstrating that these complexes could serve as autoantigens that activate B cells.24 Therefore, NETs are the main source of autoantigens (e.g. histones, LL-37 a human neutrophil antimicrobial peptide, and self-DNA) in SLE and a powerful complement activator. Serum from patients with active SLE demonstrated a reduced ability to degrade NETs in vitro. These patients had low levels of complement C3 and C4 in blood. In addition, autoantibodies against NETs inhibited their degradation and increased C1q deposition, thus exacerbating the disease.25 The high anti-dsDNA and anti-nuclear autoantibody concentration were related to the increased release of NETs in antiphospholipid syndrome and SLE.26
Hakkim et al.,27 in turn, showed that the DNase-1 serum endonuclease is key to the disassembly of NETs. Impairment of DNase-1 function and the altered degradation of NETs were associated with low levels of C3 and C4, high activity of IFN-I, high levels of autoantibodies against NETs, disease activity,25 and renal damage.27 Another study showed that NETs and LL-37 activate caspase-1, the main enzyme of the inflammasome, cause IL-1β and IL-18 production. Inflammasome activation increases in macrophages derived from SLE patients.28 This leads to the secretion of inflammatory cytokines that encourage damage to the organs. NETs generate antibody production by memory B cells through LL37-DNA complexes. These complexes enter the endosomal compartments of B cells and activate TLR9. In SLE patients, LL37-DNA complexes trigger the TLR9-dependent polyclonal activation of B cells and the expansion of autoreactive memory B cells that produce anti-LL37 autoantibodies.29
A recent study by Safi et al.,30 detected NETs in different subtypes of cutaneous lupus (lupus panniculitis (49%), acute cutaneous SLE (41%), and SLE (32%)). In addition, NETs are associated with vasculopathy and may promote coronary plaque formation and lipoprotein deregulation in patients with SLE.31 Enzymes derived from NETs, which include MPO, NADPH oxidase (NOX), and nitric oxide synthase oxidize high-density lipoprotein making it proatrogenic in SLE.32 Plasma NET levels identified the patients with severe SLE phenotypes, high SLEDAI risk, arterial events, and endothelial cell activation.33 NETs produced ex vivo are composed of proteins which have a profile that allows the characterization of SLE subphenotypes (i.e. methyl oxidized α-enolase is overexpressed in lupus nephritis).34 Moreover, in lupus nephritis, NETs are a source of extracellular high mobility group box-1 protein, which correlates with the histopathological and clinical features of the disease.35
Markers associated with NETs have been described in drug-naïve SLE patients. Jeremic et al.36 showed that serum from SLE patients has a decreased NETolytic activity, which leads to an increase in the levels of markers related to NETs (BAFF and MPO). These markers correlate with anti-dsDNA antibody levels. Human neutrophil peptides (HNP) are part of the NETs, and their impaired degradation is related to lupus nephritis. In addition, serum levels of HNP1-3 correlated with urinary protein excretion and disease activity.37 Neutrophils of patients with SLE showed a decrease in receptor-interacting protein kinase-1 (RIPK1) expression. This shows that RIPK1 interacts with protein kinase-1 and may be involved in neutrophil death and NET formation.38 Moreover, a study has shown that the binding of the signal inhibitor receptor in leukocytes-1, a negative regulator of neutrophil function, prevents the pathogenic release of NETs in SLE.39
Bone marrow cell cDNA microarrays from active SLE patients demonstrate an apoptosis and granulopoiesis signature.40 The blood transcriptome of pediatric SLE patients demonstrates an increase in neutrophil transcripts in the course of progression to lupus nephritis.41,42 Epigenetic studies revealed that an inflammatory subset of neutrophils (low-density granulocytes) have a DNA demethylation of interferon signature genes.43 Autophagy is a fundamental NET release mechanism. Peripheral blood neutrophils from SLE patients have higher levels of autophagy than neutrophils of patients with inactive SLE or healthy individuals. Autophagy inhibitors were able to attenuate the release of NETs in neutrophils of patients with active SLE.44 Mitochondrial DNA (mtDNA), in turn, has been found in NETs of patients with SLE. In addition, anti-mtDNA antibodies were significantly correlated with the IFN scores, disease activity index, and lupus nephritis.45 Some studies show that high concentrations of circulating cell-free DNA in plasma may be associated with abnormal regulation of NETs and lupus nephritis.36,46
NETs involve the covalent modification of histones, which organize chromatin within the NETs. Modification of histones by peptidylarginine deiminase (PAD) 4 is essential for the release of NETs.47 The histones present in NETs of patients with SLE show an increase in acetylated and methylated residues associated with apoptosis. Neutrophil treatment with histone deacetylase inhibitor induces NETs with hyperacetylated histones and greater capacity to activate macrophages.48 Apoptotic microparticles with acetylated chromatin induce the release of NETs independent from the formation of reactive oxygen species in patients with SLE which can lead to glomerular deposition of NETs and subsequent lupus nephritis.49 The ubiquitinated proteins present in NETs modify the cellular response and induce inflammatory mechanisms in patients with SLE. Patients with SLE develop anti-ubiquitinated MPO antibodies, and their titers correlate positively with disease activity and negatively with complement components.50
Vitamin D therapy in patients with SLE and low levels of vitamin D prevents endothelial damage by decreasing NET formation.51 Therapeutic intervention with Rituximab+Belimumab decreased the excessive formation of NETs and improved the disease through the decrease of antinuclear autoantibodies.52 In murine models of SLE, the inhibition of PAD reduces the formation of NETs and therefore protects against vascular, renal, and cutaneous damage.53,54 A reduced ability to form NETs was observed in mice with pristane-induced lupus and NOX2 and PAD4 deficiency. In addition, the mice had high levels of anti-nuclear autoantibodies and glomerulonephritis.55
NETs in rheumatoid arthritis (RA)In RA, neutrophils are the most abundant cells in the synovium and are highly activated, which are characterized by their extended survival and the production of large amounts of cytokines and chemokines. Through these mediators, neutrophils control the function of other immune cells, like monocytes, natural killer (NK) cells, DCs, etc.56
Neutrophils in RA supply citrullinated proteins through different mechanisms. Autoantibodies against citrullinated proteins or peptides have, so far, been the most sensitive and specific disease markers of RA. Therefore, they are used as both diagnostic tests and a predictive tool for RA.57 Anti-citrullinated protein/peptide antibodies (ACPA) recognize the amino acid citrulline, which is the deiminated form of arginine residue, within protein sequences.58 Although citrullination is a physiological process, its upregulation during inflammation causes organ damage as in the case of RA joints. In patients with RA, proteins such as fibrinogen, filaggrin, collagen II, vimentin, and histones are the targets of ACPA. All these proteins become the focus of ACPA after being post-translationally modified by the calcium-dependent enzyme PAD. This enzyme deiminates, a process also known as citrullination.59 Khandpur et al.60 showed that neutrophils from the peripheral blood (PB) and synovial fluid (SF) of patients with RA form more NETs compared to the neutrophils from PB taken from the control group or to neutrophils from SF of osteoarthritis patients without stimuli. Moreover, they showed that netting neutrophils were present as infiltrating cells in synovial fluid, rheumatoid nodules, and the skin of RA patients.
These netting neutrophils release active PAD2 and PAD4 which citrullinate proteins. Moreover, single nucleotide polymorphisms in PADI2 and PADI4 have been associated with RA.61,62 The precise role of each isoform in RA is still unknown although pan-PAD inhibitors reduce collagen induced arthritis.63 Several studies in RA-murine models have shown the contribution of PAD4 to inflammation64–66 although, in the TNF-α induced arthritis model where PAD4 is absent, no reduction of protein citrullination in the lungs67 or serum64 was observed. In contrast, PAD2 activity is highly correlated with disease activity, overall PAD activity, and more important with PAD2 levels in SF. However, Bawadekar et al.68 showed that although PAD2 induces TNF-α citrullination and arthritis, it is not requisite for NETosis. In addition to PADs, there are many other proteins contained in the NETs. A recent proteomic analysis of NETs in SF drawn from patients with RA showed the presence of matrix metalloproteinases (MMP)9, MMP8, MPO, lipocalin 2, and cathepsins, which are responsible for collagen degradation within synovial joints.69 Some of these proteins have already been used as biomarkers, as is the case of MMP8, which predicts RA mortality.70 Many citrullinated proteins have been detected in sera and SF of RA patients, and are clearly known as sources of autoantigens. This is the case with citrullinated vimentin that is the antigen of anti-Sa auto-antibodies.71 Moreover, citrulline residues in vimentin and aggrecan are recognized by antigen-presenting cells in individuals with the HLA-DRB1*04:01/04 allele, which contains the “shared epitope”.72 Citrullinated peptides within NETs are engulfed by synovial fibroblasts through the RAGE-TLR9 pathway to then be presented to antigen-specific T-cells via MHC Class II.73 Citrullinated histones are also a source of autoantigens. The citrullinated histones H2A, H2B and H4 have been identified in NETs.74 Moreover, B cell clones from SF of RA patients produce antibodies which strongly react with these citrullinated histones.75
NETosis is a physiological process. Whether the disbalance in RA is due to enhanced NETosis, increased extrusion of NETs or ineffective clearance is not clearly known. Pérez-Sánchez et al.76 showed that sera from RA patients had reduced DNase-I activity, which resulted in diminished DNA degradation. The authors suggested that this activity reduction could help the NET accumulation, thus influencing the progress of RA and the creation of an atherothrombotic status in RA patients. Remarkably, a clinical trial showed that, in addition to preventing venous thromboembolism, aspirin can also inhibit NETosis in vitro.77 Moreover, heparin, which is extensively used as an anticoagulant disassembles NETs15 and counteracts the interactions of histone-platelet.78 Likewise, it was demonstrated that the anti-inflammatory N-acetylcysteine, ketoprofen, and ethamsylate reduced NET formation.79 Nowadays, there are biological therapies such as anti-TNF-α inhibitors and anti-IL6R inhibitors that are able to inhibit NETosis in RA.76
NETs in Sjogren's syndrome (SS)SS is an autoimmune epithelitis characterized by mononuclear cell infiltration of secretory glands (mainly salivary and lachrymal), production of autoantibodies and pro-inflammatory mediators which lead to the appearance of sicca symptoms (i.e. xerostomia and keratoconjunctivitis).80 The accessibility to nuclear antigens such as DNA and histones due to clearance defects in NETosis can induce autoantibody formation and these can induce NETs, thus leading to a vicious cycle that exacerbates the inflammation process in autoimmunity.81
Dwivedi et al.82 showed that inflammatory conditions trigger the citrullination of core histones in neutrophils by PAD4, and patients with SLE and SS have autoantibodies against citrullinated histone H1 present in NETs. Authors demonstrated that H1 has several arginine residues in its globular domain and flexible tail, and these can be citrullinated during neutrophil activation. Furthermore, mature neutrophils have at least five different H1 proteins that could be an important source for generating new autoantibodies. In addition, stimulation of H1 reactive B-lymphocytes can lead to neutrophil activation and the release of NETs in susceptible individuals.82
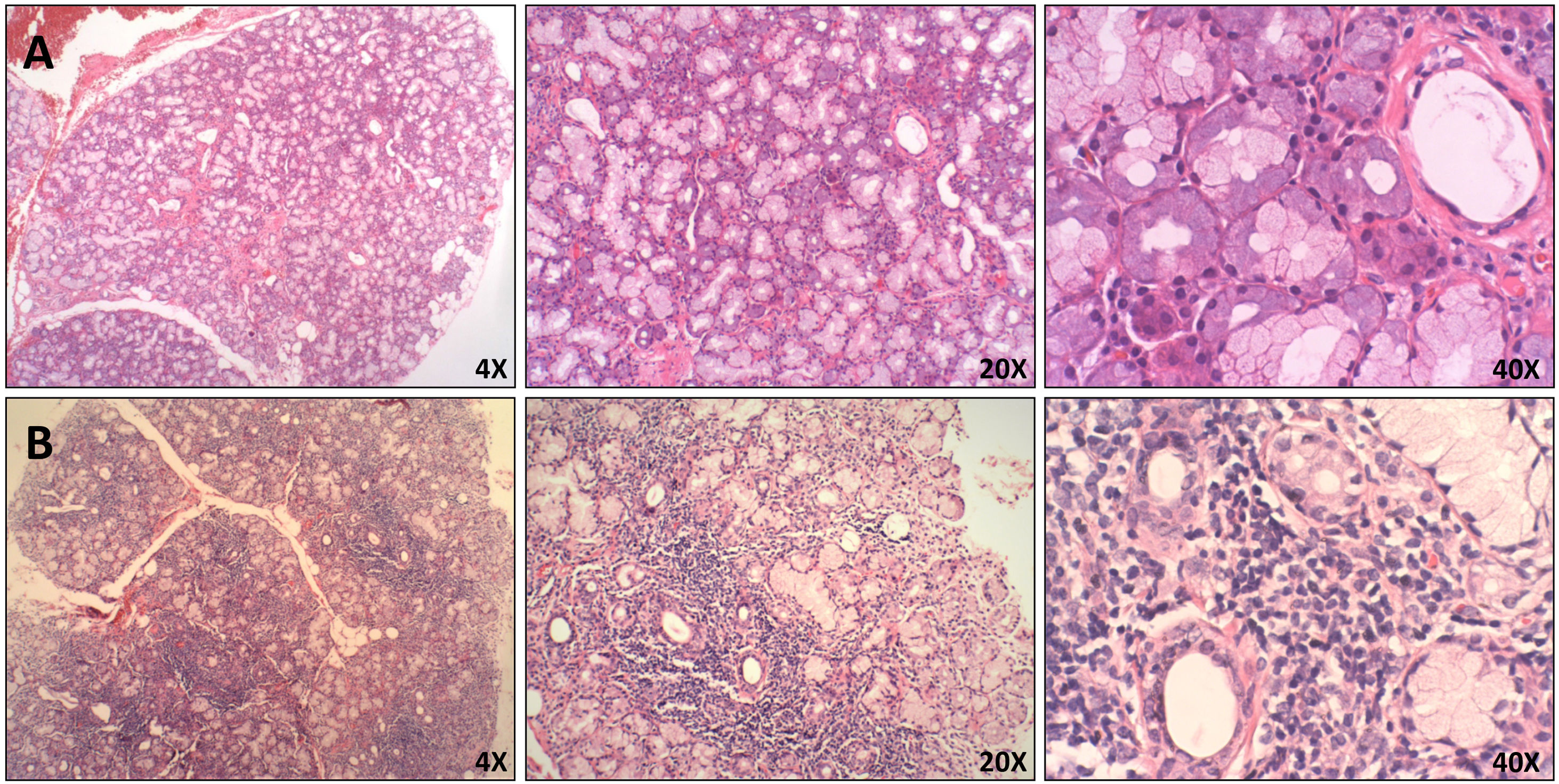
As mentioned above, NETosis has been widely studied in the pathogenesis of AAV, SLE, and RA. However, its role in SS is unclear. The study of NETs has mainly been done by its induction in vitro displayed through flow cytometry or immunofluorescence microscopy.83,84 However, a method that is ideal for evaluating the role of NET formation in vivo would be its detection on directly affected tissues. NETs in tissues are hardly ever detected by conventional stains, and until now, few articles have described a general protocol for fluorescent staining of NET components in biopsies.85,86 In addition, the use of biopsies of paraffin-embedded tissues in particular could provide optimal protection and maintenance of the structures. This would make it possible to analyze archival cytologic material and do retrospectives studies.85,87 Given the later, the presence of neutrophils and NETs in paraffin-embedded tissues from minor salivary gland biopsies (MSGB) of patients with established SS with or without polyautoimmunity were evaluated, and subjects with non-specific chronic sialadenitis were included as control group (Fig. 1). All patients in the study fulfilled the American College of Rheumatology/European League Against Rheumatic Disease (ACR/EULAR) classification criteria for SS. These criteria include clinical and serological findings and abnormalities in histologic examination. The positivity of anti-Ro antibodies and the presence of focal lymphocytic sialadenitis are the major criteria.88 Polyautoimmunity was considered in patients that fulfill the criteria for more than one autoimmune disorder.89

Histopathological features of MSGB. Hematoxilin and eosin staining of minor salivary glands from A. Patient with non-specific chronic sialadenitis characterized by focal or scattered cell infiltrate adjacent to apparently normal acini and B. Patient with SS characterized by focal lymphocytic sialadenitis defined as the presence of ≥50 mononuclear cells in perivascular or periductal areas.
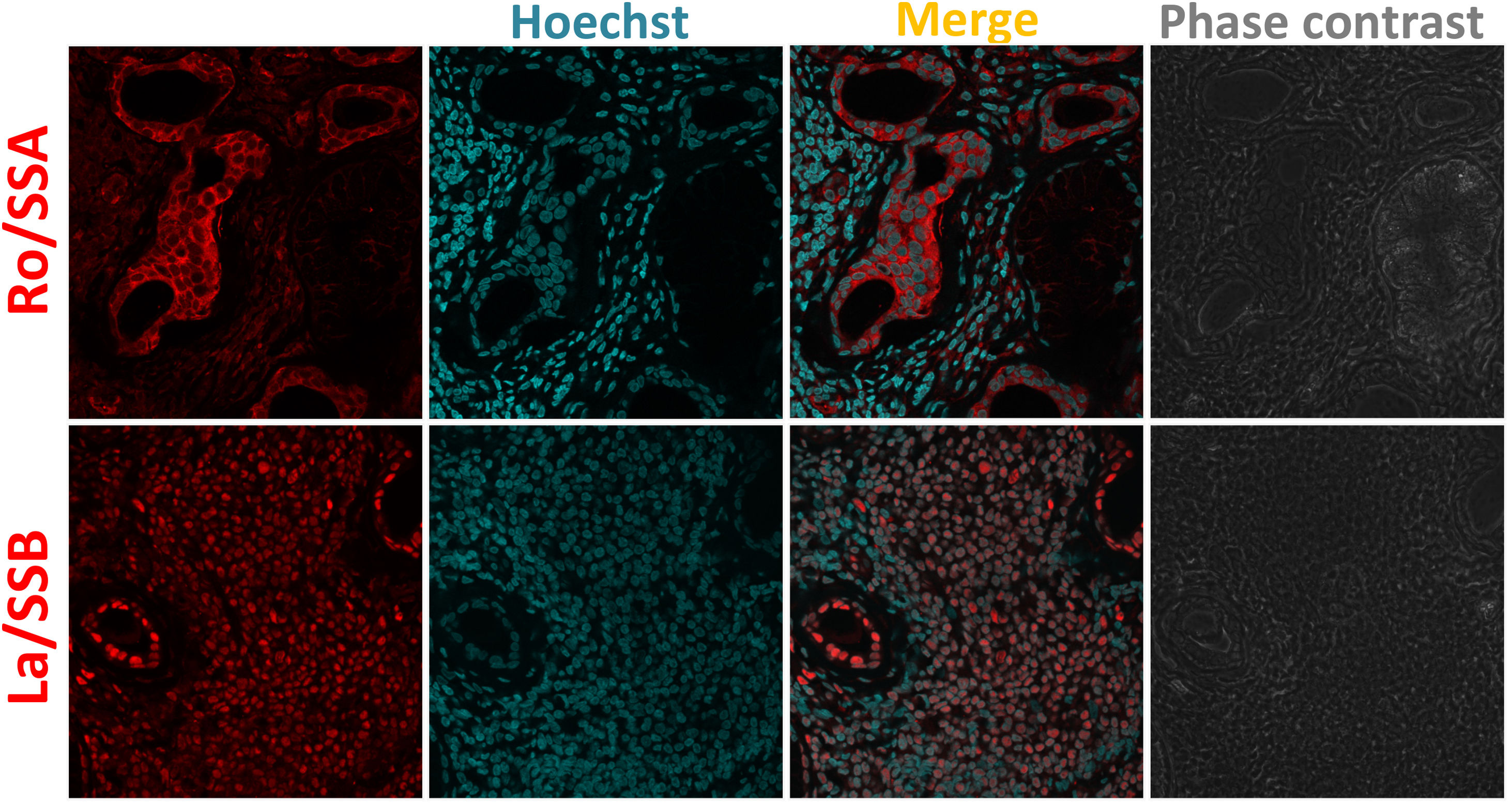
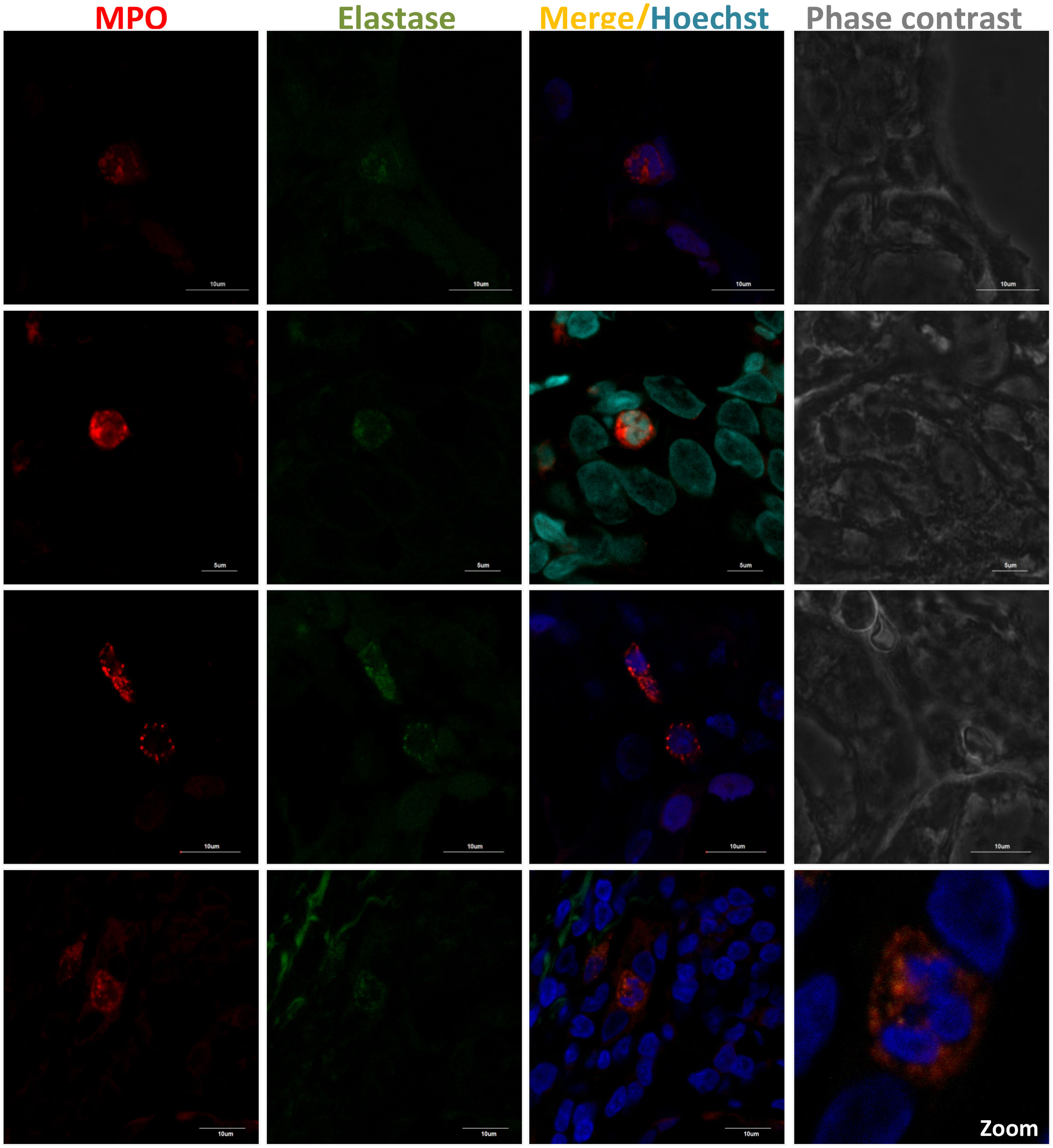
MSGB plays an important role in the diagnosis and stratification of SS.90 The presence of 50 or more mononuclear cells located in periductal or perivascular zones is considered one focus. When at least one focus is observed, calculation of the focus score (F.S) is necessary.91 An FS equal to or greater than one (≥1) is considered positive. This feature is one of the five classification criteria for SS established by ACR/EULAR.88 In order to detect neutrophils or NETs in these biopsies, the standardization of several conditions for a successful fixation, dehydration, embedding into paraffin, rehydration, antigen retrieval, and immunofluorescent staining was done (supplementary table S1). By utilizing double sequential immunofluorescent staining, we detected Ro/SS-A and La/SS-B, the most common autoantigens associated with the pathogenesis of SS92 (Fig. 2). In addition, markers for neutrophils were detected in MSGB from patients with FS ≥1 in contrast with the control group (Fig. 3). However, the neutrophil traps were not displayed. Histopathological detection of NETs is not easy. One of the main pathogenic aspects of this chronic disease is the nature of its cell infiltration in periductal or perivascular (around epithelial cells) areas and the hyperactivity of these infiltrating cells.93
 and rabbit monoclonal anti-La/SSB (ab124932). Secondary fluorescent antibody was goat anti-rabbit IgG alexa fluor 546 (red) mixed with Hoechst (blue) used to stain nucleic acids. The subcellular location of Ro/SSA antigen is nuclear and cytoplasmic. Ro/SSA is mainly found in the cytoplasm, whereas La/SSB is observed in nuclei.")
Ro/SSA and La/SSB subcellular location in MSGB by indirect immunofluorescence. 4um-thick MSGB sections were tested with rabbit polyclonal anti-Ro/SSA/TRIM21 (ab91423) and rabbit monoclonal anti-La/SSB (ab124932). Secondary fluorescent antibody was goat anti-rabbit IgG alexa fluor 546 (red) mixed with Hoechst (blue) used to stain nucleic acids. The subcellular location of Ro/SSA antigen is nuclear and cytoplasmic. Ro/SSA is mainly found in the cytoplasm, whereas La/SSB is observed in nuclei.
 with elastase (green) was detected using rabbit polyclonal anti-MPO (ab9535) and mouse monoclonal anti-neutrophil elastase (sc-55549). Secondary fluorescent antibodies were goat anti-rabbit IgG alexa fluor 546 (A-11010) and goat anti-mouse IgG alexa fluor 488 (A-11001) mixed with Hoechst or DAPI (blue) used to stain nucleic acids.")
Presence of neutrophils in MSGB from SS patients with FS ≥1. NET components in 4μm-thick MSGB sections were evaluated by double sequential immunofluorescence. Colocalization of MPO (red) with elastase (green) was detected using rabbit polyclonal anti-MPO (ab9535) and mouse monoclonal anti-neutrophil elastase (sc-55549). Secondary fluorescent antibodies were goat anti-rabbit IgG alexa fluor 546 (A-11010) and goat anti-mouse IgG alexa fluor 488 (A-11001) mixed with Hoechst or DAPI (blue) used to stain nucleic acids.
Several studies have shown that the main types of inflammatory cells in SS patients’ MSGB are T and B cells (∼90%), whereas macrophages, DCs, and NK cells are a small proportion (∼5–10%).94–96 The incidence of these cells can vary based on the severity of the lesion. For instance, T cells prevail in mild lesions in contrast with B cells that predominate in advanced ones.93,95 Although neutrophils are unlikely to be found in these biopsies, these cells were seen in MSGB from patients with FS≥1 and correlated with the local immunopathological reaction. A study of the morphology of the MSG in SS showed that a strong and active inflammatory reaction included the presence of T cells, macrophages and neutrophils, an increase in vascular permeability, and changes in intralobular ducts and acini. However, in reduced inflammatory activity, the quantity of macrophages and neutrophils decreases, plasma and mast cells increase, and there is higher compensatory hypertrophy of the serous cells of mixed acini.97
Furthermore, it is difficult to not only find neutrophils but also capture the exact moment when they release their networks. Moreover, a minimum of 4 glands need to be evaluated and multiple cutting levels must be inspected to insure a reading of an adequate area and increase the probability of finding neutrophils. In spite of the fact that NETs were not detected, we found that neutrophils may be an important source of autoantigens that induce an immune response, thus leading to the development and continuance of this AD. Up to now, there are no studies that provide insights into the mechanism of NET formation in SS, thus, it is pivotal to evaluate their presence through further research in order to elucidate the possible role of neutrophils in the pathophysiology of this disease.
ConclusionThe specific role of neutrophils in the pathogenesis of the ADs is poorly understood. However, in the last few years, more and more researchers have focused on neutrophil biology and its contribution to the development and progression of ADs. Specifically, the formation of NETs is implicated in the innate immune response to several pathogens or may be produced through non-infectious stimuli (i.e. cytokines, autoantibodies or immune complexes). However, in acute or chronic disorders an uncontrolled NET formation and their defective removal are associated with autoantigen exposition, development of disease-specific autoantibodies, and inflammation perpetuation, which can be closely associated with the development of ADs such as AAV, SLE, and RA. However, in SS this mechanism and its effect still are unknown. Further studies are needed to assess whether NET components could ease diagnosis, be used as biomarkers in ADs, or be novel therapeutic targets for the treatment of these diseases without altering host defense.
FundingThis work was supported by ASOREUMA (convocatoria pública para proyectos de investigación 2016 – Asociación Colombiana de Reumatología) and Universidad del Rosario (ABN011).
Conflict of interestAll authors state that they have no conflict of interest.
We would like to express our gratitude to Yovana Pacheco and Adriana Rojas-Villarraga for their contribution to this work.
The following are the supplementary data to this article:
