



La poliautoinmunidad se define como la presencia de más de una enfermedad autoinmune (EA) bien caracterizada en un mismo paciente. Es una condición frecuente en pacientes con síndrome de Sjögren (SS) y sigue un patrón de agrupamiento. Las EA más frecuentes observadas en el SS son la enfermedad tiroidea autoinmune, la artritis reumatoide y el lupus eritematoso sistémico. El estudio de este fenómeno aporta claves importantes para entender los mecanismos comunes de las EA.
Multiple autoimmunity is defined as the presence of more than one well-defined autoimmune disease (AD) in a single patient. Multiple autoimmunity is a frequent condition in Sjögren's syndrome (SS) and follows a grouping pattern. The most frequent ADs observed in SS are autoimmune thyroid disease, rheumatoid arthritis, and systemic lupus erythematosus. The study of multiple autoimmunity provides important clues for elucidating the common mechanisms of ADs.
Las enfermedades autoinmunes (EA) son condiciones crónicas que resultan de la pérdida de la tolerancia inmunológica a antígenos propios debido a una compleja interacción entre factores hereditarios y medioambientales a lo largo del tiempo1. Diversos argumentos apoyan un origen común de las EA, entre ellos la coexistencia de 2o más enfermedades en un mismo individuo (p. ej., poliautoinmunidad)2,3.
El síndrome de Sjögren (SS) es una EA sistémica heterogénea. Se caracteriza por la presencia de un infiltrado linfoplasmocitario en las glándulas exocrinas, lo cual trae como resultado una disfunción glandular y los síntomas de sequedad característicos4. Debido a que el tejido diana involucrado en esta entidad corresponde al epitelio, el cual tiene un papel central en su fisiopatogenia, en la actualidad se usa el término «epitelitis autoinmune» para describir esta enfermedad5.
El espectro clínico del SS es amplio y puede extenderse desde una exocrinopatía autoinmune (manifestaciones glandulares) hasta una enfermedad con afectación sistémica (manifestaciones extraglandulares). Los mecanismos fisiopatogénicos subyacentes incluyen la producción de autoanticuerpos, especialmente anticuerpos antinucleares (ANA), anti-Ro/SS-A, anti-La/SS-B, factor reumatoide (FR) y crioglobulinas, la formación de complejos inmunes y la secreción de mediadores inflamatorios, entre otros6.
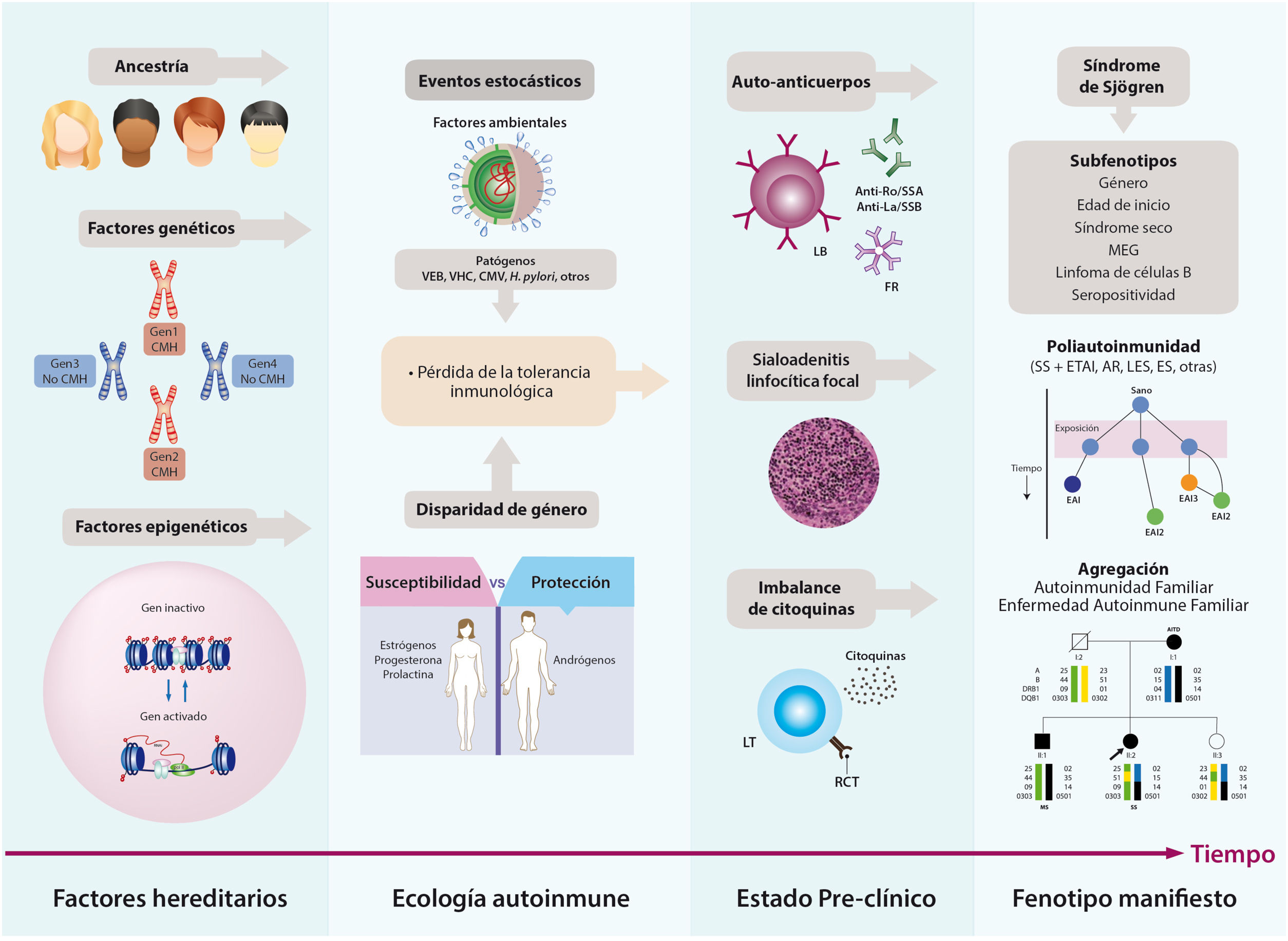
La expresión fenotípica del SS es el resultado de la pérdida de la tolerancia inmunológica, en lo cual intervienen factores genéticos, epigenéticos y ambientales. Lo anterior se traduce en la activación persistente de células autorreactivas y en la instauración de una respuesta inmunitaria humoral aberrante que trae consigo el daño tisular. Otros factores implicados son la edad de inicio, el género, las comorbilidades y la poliautoinmunidad7. Lo anterior explica en gran medida la amplia heterogeneidad de esta enfermedad (fig. 1).

Historia natural del SS. La etiología del SS es multifactorial; en ella, un mosaico de factores hereditarios y estocásticos interactúan y traen como resultado la pérdida de la tolerancia inmunológica y el subsiguiente daño orgánico. El estado preclínico se caracteriza por la presencia de autoanticuerpos en suero, el infiltrado linfoplasmocitario en glándulas salivales y la producción de mediadores solubles proinflamatorios antes de la primera manifestación de la enfermedad. La fase clínica hace referencia a la expresión fenotípica compatible con SS, la cual incluye diversas formas de presentación o subfenotipos, incluida la poliautoinmunidad.
AR: artritis reumatoide; CMH: complejo mayor de histocompatibilidad; CMV: citomegalovirus; EA: enfermedad autoinmune; ES: esclerosis sistémica; ETAI: enfermedad tiroidea autoinmune; FR: factor reumatoide; LES: lupus eritematoso sistémico; LT: linfocito T; MEG: manifestaciones extraglandulares; RCT: receptor de célula T; SS: síndrome de Sjögren; VEB: virus de Epstein-Barr; VHC: virus de la hepatitis C.
Desde la primera descripción hecha por Henrik Sjögren en 1933, es bien conocido que la mayoría de los pacientes con SS presenta poliautoinmunidad. De los 19 pacientes originalmente descritos, el 68% presentaba una artritis deformante compatible con artritis reumatoide (AR)8. En 1968 Pirofsky y Vaughn9 reportaron 5 pacientes con enfermedad de Addison, anemia perniciosa y positividad para anticuerpos antitiroglobulina, y propusieron el término «síndrome autoinmune múltiple» (SAM) para referirse a la presencia de 3o más EA en un mismo individuo. Con posterioridad, en 1988, Humbert y Dupond10 recopilaron una serie de casos de 91 pacientes con SAM y propusieron una clasificación de acuerdo con la prevalencia de las posibles asociaciones y los mecanismos patogénicos subyacentes (p. ej., predisposición genética). En tal sentido, clasificaron el SS junto con entidades como la AR, la enfermedad tiroidea autoinmune (ETAI) y el lupus eritematoso sistémico (LES), entre otras.
En 1993 Sheehan y Stanton-King11 acuñaron el término «poliautoinmunidad» para describir a una paciente con AR, púrpura trombocitopénica idiopática, anemia perniciosa, tiroiditis de Hashimoto (TH), esclerosis sistémica (ES), insuficiencia pancreática exocrina y enfermedad celíaca, que tuvo un desenlace fatal debido a complicaciones vasculíticas. Adicionalmente, describieron la presencia de AR, anemia perniciosa y diabetes mellitus tipo 1 en sus familiares de primer grado (p. ej., autoinmunidad familiar). Tanto la poliautoinmunidad como la autoinmunidad familiar son argumentos a favor de la presencia de mecanismos fisiopatogénicos comunes a las EA, por lo cual el uso de términos como «EA secundaria» podría no ser preciso y llevar a una amplia discusión taxonómica12.
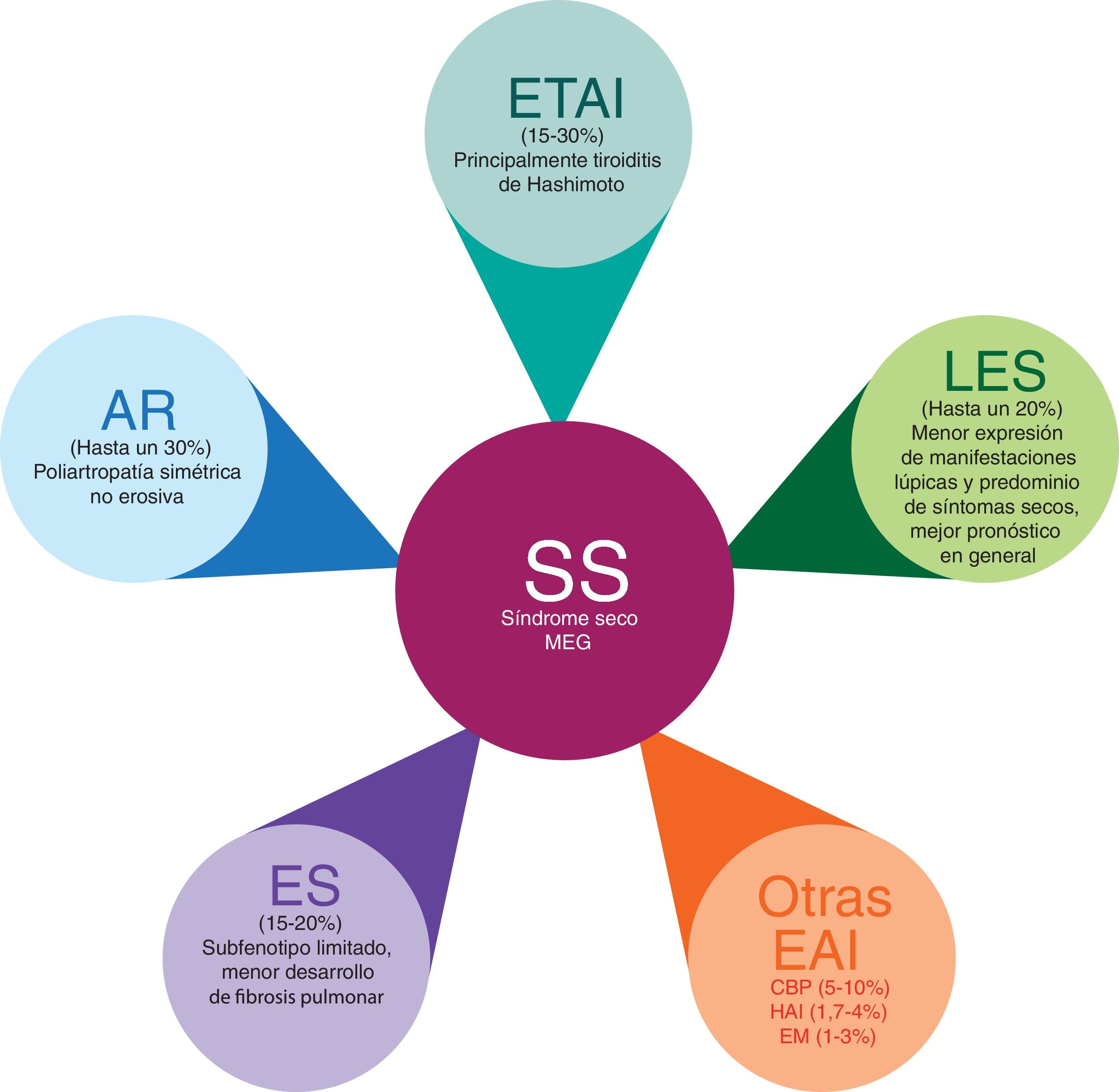
Poliautoinmunidad en síndrome de SjögrenEl SS se ha descrito en asociación con una amplia variedad de EA órgano-específicas y sistémicas (fig. 2), como la ETAI13-17, la AR18-26, el LES18,27-36, la ES32,37-40, la colangitis biliar primaria (CBP)41-45, la hepatitis autoinmune (HAI)41,46-50, entre otras51-53. La poliautoinmunidad es un fenómeno frecuente, con prevalencias reportadas hasta un 52% en pacientes con SS54. Dicha prevalencia varía según la geolocalización de la población estudiada y los criterios tenidos en cuenta para clasificar a estos pacientes55.

Poliautoinmunidad en el SS.
AR: artritis reumatoide; CBP: colangitis biliar primaria; EM: esclerosis múltiple; ES: esclerosis sistémica; ETAI: enfermedad tiroidea autoinmune; HAI: hepatitis autoinmune; LES: lupus eritematoso sistémico; MEG: manifestaciones extraglandulares; SS: síndrome de Sjögren.
Una revisión sistemática de la literatura evaluó la epidemiología de la poliautoinmunidad en pacientes con SS e incluyó aquellos estudios de buena a moderada calidad en un metaanálisis56. La tasa de prevalencia agrupada para el subgrupo SS-AR fue del 19% (IC del 95%: 11,2-27,8) y para SS-LES del 14% (IC del 95%: 8,8-19). Los estudios incluidos en el metaanálisis se caracterizaron por un alto grado de heterogeneidad (I2=99,92 y 99,98, respectivamente).
Se han reportado varios casos de SAM que incluyen el SS57-60. Las EA descritas en este contexto incluyen especialmente las hepatopatías autoinmunes (HAI-CBP), la TH, la AR y la psoriasis. Otras entidades reportadas con menos frecuencia son la enfermedad celíaca, el LES, la ES y el vitíligo. Efe et al.57 evaluaron a 71 pacientes con HAI-CBP, con el objetivo de identificar otras EA asociadas. Un 43% (31 pacientes) fue clasificado con EA extrahepáticas, principalmente ETAI (18%), SS (8%), AR (4%), enfermedad celíaca (4%) y psoriasis (4%). Se diagnosticaron un total de 181 EA, 2 de ellas presentes en un 56% de los pacientes y 3 o más en los restantes.
Amador-Patarroyo et al.61 evaluaron una cohorte de 410 pacientes colombianos con SS (criterios del Consenso Americano-Europeo 2002) y observaron una prevalencia de poliautoinmunidad del 32%, teniendo en cuenta criterios clasificatorios validados para un panel de EA. Las enfermedades coexistentes más frecuentes y estrechamente relacionadas fueron la ETAI (21%), la AR (8%) y el LES (7%). El SAM se reportó en un 8% de los pacientes. En población británica, Lazarus e Isenberg62 reportaron resultados similares.
Enfermedad tiroidea autoinmuneLos síntomas endocrinos documentados en pacientes con SS se deben principalmente a una disfunción tiroidea concomitante4. Entre 15 y 30% de los pacientes con SS desarrollan ETAI, principalmente TH13. La prevalencia de anticuerpos antitiroperoxidasa (anti-TPO) y antitiroglobulina (anti-Tg) en pacientes con SS-ETAI es del 9-45% y del 3-36%, respectivamente13. Los pacientes eutiroideos, pero seropositivos para anticuerpos antitiroideos (anti-TPO o anti-Tg) representan un subgrupo particular que puede progresar a hipotiroidismo clínico a lo largo del tiempo63. Por lo tanto, es necesario un seguimiento regular de dicha población en riesgo64. El SS puede preceder a la TH hasta en 5 años en el 50% de los casos65.
La prevalencia de SS es 10 veces mayor en pacientes con ETAI14. Un tercio de los pacientes con ETAI tiene características de SS y uno de cada 10 pacientes con ETAI y ANA positivos comparte el diagnóstico de SS66. Es recomendable evaluar sistemáticamente la función tiroidea en pacientes con SS, con el objetivo de identificar a pacientes tributarios de tratamiento hormonal suplementario14.
Los hallazgos histopatológicos en la TH (infiltrado linfoplasmocitario) son muy similares a los descritos en el SS5. La expresión clínica del SS varía ligeramente cuando coexiste la TH. La asociación SS-TH define un subgrupo de pacientes con una enfermedad más benigna y con menor prevalencia de factores de riesgo de desarrollo de linfoma (p. ej., niveles normales de C4)15.
Anaya et al.16 llevaron a cabo un estudio retrospectivo de un solo centro en el que incluyeron 293 pacientes colombianos con SS (criterios ACR/EULAR 2016). Los investigadores compararon a pacientes con SS primario y aquellos con SS-ETAI. Todos los pacientes con ETAI presentaban TH. Los pacientes del subgrupo SS-TH presentaron con mayor frecuencia linfadenopatías y urticaria, al igual que historia de tabaquismo. Aunque ambas entidades comparten mecanismos fisiopatológicos comunes, son nosológicamente diferentes y su coexistencia debe interpretarse como poliautoinmunidad16.
Artritis reumatoideLa afectación articular representa una de las manifestaciones sistémicas más frecuentes en el SS4. Se ha estimado que entre el 37-75% y el 11-22% de los pacientes con SS presenta artralgias y artritis, respectivamente67,68. La artritis suele ser de distribución poliarticular, simétrica y no erosiva69.
Por otro lado, los síntomas secos hacen parte del espectro de manifestaciones extraarticulares de la AR. Lo anterior indica que los pacientes con AR pueden presentar ojo seco, con independencia de la coexistencia de SS70. La prevalencia de síntomas secos en pacientes con AR varía del 30 al 50%18,19 y el porcentaje de pacientes con AR que cumplen criterios de clasificación de SS se ha reportado entre el 4 y el 31%18-20,22,24,71. Algunos autores consideran que el SS es una manifestación sistémica de la AR, pese a que se han documentado diferencias fisiopatogénicas involucradas en el desarrollo de cada entidad18.
La prevalencia de los anticuerpos antipéptidos cíclicos citrulinados (ACPA) en el SS se encuentra entre el 3 y el 10%72. El seguimiento de los pacientes con ACPA positivos y SS mostró que casi la mitad de ellos desarrollaron AR73. En pacientes colombianos, se observaron ACPA en el 9% de los pacientes con SS, de los cuales un 80% eran FR positivos. Esta prevalencia fue mayor (67%) en pacientes con SS-AR74. Iwamoto et al.75 detectaron ACPA en el 21% de los pacientes con SS con artritis y en ninguno de los que no presentaban artritis. En particular, se encontraron ACPA en el 71% de los pacientes clasificados con SS-AR y en el 6% de aquellos con SS y artritis, pero sin criterios de AR. El FR no fue útil para diferenciar entre pacientes con SS de aquellos con SS-AR75.
Molano-González et al.72 llevaron a cabo un metaanálisis con el objetivo de evaluar la relación entre la presencia de artritis y la seropositividad para ACPA en SS primario, al igual que su papel en el desarrollo de AR. A tal efecto, incluyeron a 1.322 pacientes y encontraron una asociación (OR 4,42; IC del 95%: 1,15-16,94; p=0,03) entre la artritis y los ACPA. El 57% (33/58) de los pacientes ACPA positivos desarrolló AR, en comparación con ninguno de los 598 pacientes seronegativos (p<0,000001). Se ha descrito con mayor frecuencia afectación pulmonar en pacientes con SS y ACPA positivos73. Los pacientes con SS-AR son menos frecuentemente seropositivos para anticuerpos anti-Ro/SS-A y anti-La/SS-B que los pacientes con SS primario (12% vs. 82%)76.
El número de articulaciones afectadas y la actividad de la AR es independiente de la presencia de SS77. En población de la India, los pacientes con SS-AR tienden a experimentar una mayor duración de la enfermedad, al igual que una mayor edad, en comparación con aquellos con SS primario. Sin embargo, no se encontró diferencia en el daño articular25. Un ensayo clínico multicéntrico demostró efectividad del abatacept en el dominio glandular (cambio en test de Schirmer y Saxon) y articular (tasa de remisión a las 52 semanas según SDAI), en pacientes con SS-AR78.
Yang et al.26 analizaron retrospectivamente a pacientes con SS-AR, SS y AR. Los pacientes con SS-AR tuvieron mayor compromiso visceral, incluyendo enfermedad pulmonar intersticial difusa (EPID) y alteraciones hematológicas, al igual que un mayor requerimiento de glucocorticoides, en comparación con los controles. También hubo diferencias clínicas al estratificar la población según la forma de inicio (inicio en forma de AR, SS-AR o SS). En el análisis multivariado, la presencia de artritis, xerostomía, EPID y la seropositividad para FR, ACPA o anti-Ro/SS-A fueron factores asociados de manera independiente con el subgrupo SS-AR. Por otro lado, algunos estudios han descrito una tasa de incidencia estandarizada duplicada para linfoma no Hodgkin en pacientes con SS-AR en comparación con pacientes con AR23. Todo lo anterior sugiere una mayor morbimortalidad de este subfenotipo particular.
Lupus eritematoso sistémicoEl LES es probablemente la EA más estrechamente relacionada con el SS debido a la superposición significativa en su expresión clínica e inmunológica. Ambas entidades tienden a agregarse dentro de un mismo núcleo familiar79. Los análisis del transcriptoma de células mononucleares de pacientes con LES han mostrado una expresión aumentada de genes inducidos por interferón (IFN, tipo I), conocida en la literatura como la «firma del IFN». Más recientemente, se han documentado los mismos hallazgos en glándulas salivales y células mononucleadas de pacientes con SS80. Lo anterior trae consigo el desarrollo y estudio de nuevas terapias dirigidas a esta vía patogénica.
La prevalencia del síndrome seco se ha reportado hasta en un 34% de los pacientes con LES71. En la cohorte del Grupo Latinoamericano de Estudio del Lupus (GLADEL), la cual incluyó a 1.214 pacientes con LES, el 8% de los pacientes presentó síndrome seco81. En la mayoría de casos, el LES precede al inicio del SS30. Varios estudios han descrito una prevalencia de SS en pacientes con LES que oscila entre el 9 y el 19%18,27,29,31.
Los pacientes con SS-LES representan un subfenotipo en el cual predominan las características relacionadas con el SS sobre las asociadas con el LES18,27. Algunas series han mostrado una mayor prevalencia de fatiga, manifestaciones cutáneas (p. ej., fotosensibilidad, erupción malar, úlceras orales), artritis, fenómeno de Raynaud, psicosis, trombocitopenia y anticuerpos anti-Ro/SS-A y anti-La/SS-B en pacientes con SS-LES27,30. Adicionalmente, los pacientes con SS-LES tienen una mayor edad, un menor riesgo de desarrollar glomerulonefritis y linfadenopatías, al igual que una menor presencia de anticuerpos anti-ADNdc y anti-RNP, en comparación con los pacientes con LES30.
En términos de tratamiento, se han evaluado la eficacia y la seguridad del rituximab en pacientes con LES o SS y trombocitopenia refractaria. Todos los pacientes con SS tuvieron una respuesta completa al tratamiento82. En general, el subgrupo SS-LES parece caracterizarse por una menor afectación visceral, un perfil de autoanticuerpos más específico y un desenlace clínico favorable83.
Esclerosis sistémicaLos síntomas secos son frecuentes en la ES debido a los cambios fibróticos de las glándulas salivales, con una prevalencia reportada entre el 68 y el 83% de los casos. Sin embargo, solamente el 14% de estos pacientes cumple criterios clasificatorios para SS38. Avouac et al.38 evaluaron la prevalencia del síndrome seco y el SS en una serie prospectiva multicéntrica de 133 pacientes hospitalizados con ES. El 68% de los pacientes presentó síntomas secos, en tanto que el 14% se clasificó como SS. La afectación cutánea limitada de ES y la positividad para anticuerpos anticentrómero se asociaron significativamente a este subgrupo de pacientes.
Otros estudios han descrito también que la forma limitada de ES resulta ser la reportada con mayor frecuencia en este subgrupo83. En los pacientes con SS-ES, la afectación cutánea parece ser menos grave que en aquellos con ES37,38. Asimismo, la incidencia de úlceras digitales y de hipertensión pulmonar tiende a ser menor (el 11 y el 23%, respectivamente)40. Algunos estudios sugieren que el SS puede actuar como factor protector en el desarrollo de fibrosis pulmonar asociada a ES83.
Hepatopatías autoinmunesLa afectación hepática fue una de las primeras manifestaciones sistémicas reportadas en SS46. Después de eliminar los fármacos hepatotóxicos y la enfermedad por hígado graso, las 2causas principales de enfermedad hepática en el SS son las infecciones virales crónicas y las enfermedades hepáticas autoinmunes. Cada etiología requiere un enfoque terapéutico específico y presenta un pronóstico diferente41. Con respecto a las infecciones virales, la infección crónica por virus de la hepatitis C es la principal causa de afectación hepática en pacientes con SS del área mediterránea, mientras que la infección crónica por virus de la hepatitis B puede ser la principal causa de afectación hepática en pacientes con SS de países asiáticos84.
Después de las hepatitis virales, la CBP es la principal causa de enfermedad hepática en el SS84. Los pacientes con SS-CBP pueden tener un amplio espectro de anormalidades del hígado. Desde el punto de vista histopatológico, la incidencia de colangitis no supurativa linfoide fue mayor en el grupo de pacientes SS-CBP en comparación con pacientes con CBP42. Se ha sugerido que los pacientes con SS-CBP tienen un peor pronóstico debido a un mayor riesgo de EPID y peritonitis bacteriana espontánea43.
La HAI, diagnosticada en el 1,7-4% de los pacientes con SS47, representa la segunda enfermedad hepática autoinmune observada con mayor frecuencia en pacientes con SS. Todos los casos informados son de tipo I y casi el 10% de estos pacientes pueden tener coexistencia de HAI-CBP48. En un estudio que evaluó la presencia de poliautoinmunidad en pacientes con HAI, el SS fue la EA más frecuente en un 15%49. Cuando se evaluó la prevalencia de EA extrahepática concurrente en pacientes con HAI-CBP, se observó el SS en un 8%57.
Esclerosis múltipleEntre un 15 y un 25% de los pacientes con SS puede presentar afectación del sistema nervioso central85,86. Dicha afectación es variable e incluye el síndrome similar a la esclerosis múltiple (EM) y trastornos del espectro de neuromielitis óptica. La aparición de los síntomas puede darse de forma aguda, insidiosa o recurrente, como ocurre en los pacientes con EM, considerada la gran simuladora del compromiso neurológico central en el SS87.
No existe un consenso sobre la prevalencia del SS en pacientes con EM debido a la heterogeneidad de los criterios clasificatorios y al tamaño limitado de los diferentes estudios. Pese a esto, se ha estimado la prevalencia de SS-EM entre el 0,9 y el 16%88. La EM puede aparecer durante el curso del SS, al inicio de la enfermedad o incluso precederla por varios años. Asimismo, los pacientes con EM y curso progresivo pueden presentar síntomas secos sin llegar a desarrollar un fenotipo bien definido de SS (p. ej., no llegan a cumplir criterios clasificatorios). Por tanto, el diagnóstico diferencial entre el SS y la EM puede representar un gran reto, aun para clínicos experimentados87.
ConclusionesEl SS se ha descrito en asociación con una amplia variedad de EA órgano-específicas y sistémicas. La poliautoinmunidad es una condición frecuente en pacientes con SS, por lo que su búsqueda y evaluación se debe realizar sistemáticamente. Su prevalencia varía según la geolocalización de la población estudiada y los criterios clasificatorios utilizados. Las EA más frecuentemente asociadas con el SS son la TH, la AR y el LES. La coexistencia con dichas entidades define un subfenotipo particular con influencia en la expresión fenotípica de la enfermedad, así como en su tratamiento y en el pronóstico, en la mayoría de los casos favorable.
FinanciaciónLa presente investigación no ha recibido ninguna beca específica de agencias de los sectores público, comercial o sin ánimo de lucro.
Conflicto de interesesLos autores declaran que no tienen ningún conflicto de intereses en el presente trabajo.
